nf-core/neutronstar
De novo assembly pipeline for 10X linked-reads using Supernova
This pipeline is archived and no longer maintained.
Archived pipelines can still be used, but may be outdated and will not receive bugfixes.
22.10.6.
Learn more.
Introduction
nf-core/neutronstar is a bioinformatics best-practice analysis pipeline used for de-novo assembly and quality-control of 10x Genomics Chromium data. The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible.
Quick Start
i. Install nextflow
ii. Install one of docker, singularity or conda
iii. Download the pipeline and test it on a minimal dataset with a single command
nextflow run nf-core/neutronstar -profile test,<docker/singularity/conda>iv. Start running your own analysis!
nextflow run nf-core/neutronstar -profile <docker/singularity/conda> --id assembly_id --fastqs fastq_path --genomesize 1000000See usage docs for all of the available options when running the pipeline.
Disclaimer
This software is in no way affiliated with nor endorsed by 10x Genomics.
Pipeline overview
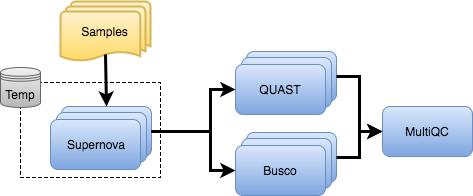
Credits
nf-core/neutronstar was originally written by Remi-Andre Olsen (@remiolsen).
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on Slack (you can join with this invite).
Citation
If you use nf-core/neutronstar for your analysis, please cite it using the following doi:
You can cite the nf-core pre-print as follows:
Ewels PA, Peltzer A, Fillinger S, Alneberg JA, Patel H, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. nf-core: Community curated bioinformatics pipelines. bioRxiv. 2019. p. 610741. doi: 10.1101/610741.