nf-core/variantcatalogue
Pipeline to generate variant catalogues, a list of variants and their frequencies in a population, from whole genome sequences.
Introduction
This document describes the output produced by the pipeline. Most of the plots are taken from the MultiQC report, which summarises results at the end of the pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
A. Mapping subworkflow
- BWA index - Index the reference genome
- Trimmomatic - Trim the reads
- BWA mem - Align the reads
- Samtools index - Index the aligned bam files
- FastQC - Raw read QC
- Mosdepth - Coverage calculation
- Picard Collect WGS metrics - Alignemnt QC
- Picard Collect Alignment Summary Metrics - Alignemnt QC
- Quality Scores distribution - Alignemnt QC
- MultiQC - Aggregate report describing results and QC from the whole pipeline
B. SNV/Indel subworkflow
- DeepVariant - Variant calling
- GLnexus - Joint calling
- Bedtools norm - Split multi-allelic variants
- Bedtools Annotate - Redefine varaints ID
- Hail Sample QC - Sample QC and sex inference
- Hail Variant QC - Variant QC and frequency calculation
- VEP - Annotation
C. Mitochondrial Subworkflow Will be included soon
D. Structural varaint subworkflow Will be included soon
- Pipeline information - Report metrics generated during the workflow execution
FastQC
Output files
fastqc/*_fastqc.html: FastQC report containing quality metrics.*_fastqc.zip: Zip archive containing the FastQC report, tab-delimited data file and plot images.
FastQC gives general quality metrics about your sequenced reads. It provides information about the quality score distribution across your reads, per base sequence content (%A/T/G/C), adapter contamination and overrepresented sequences. For further reading and documentation see the FastQC help pages.
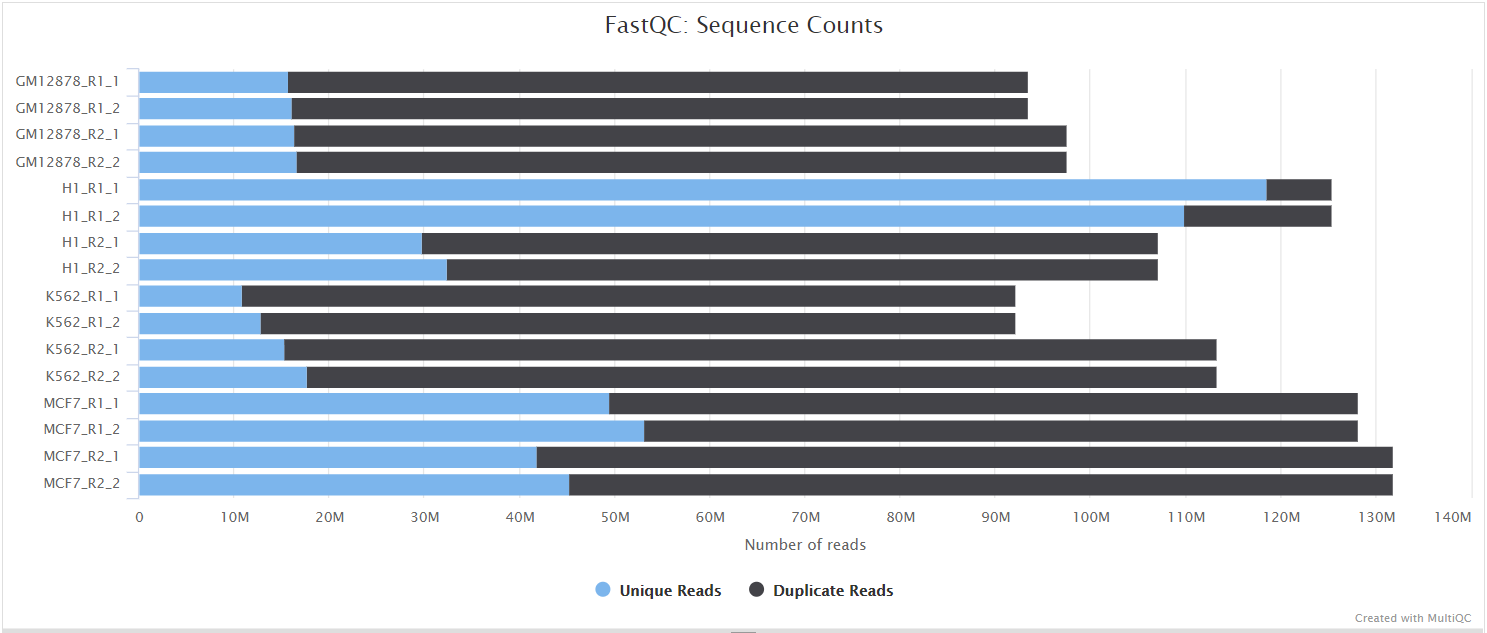
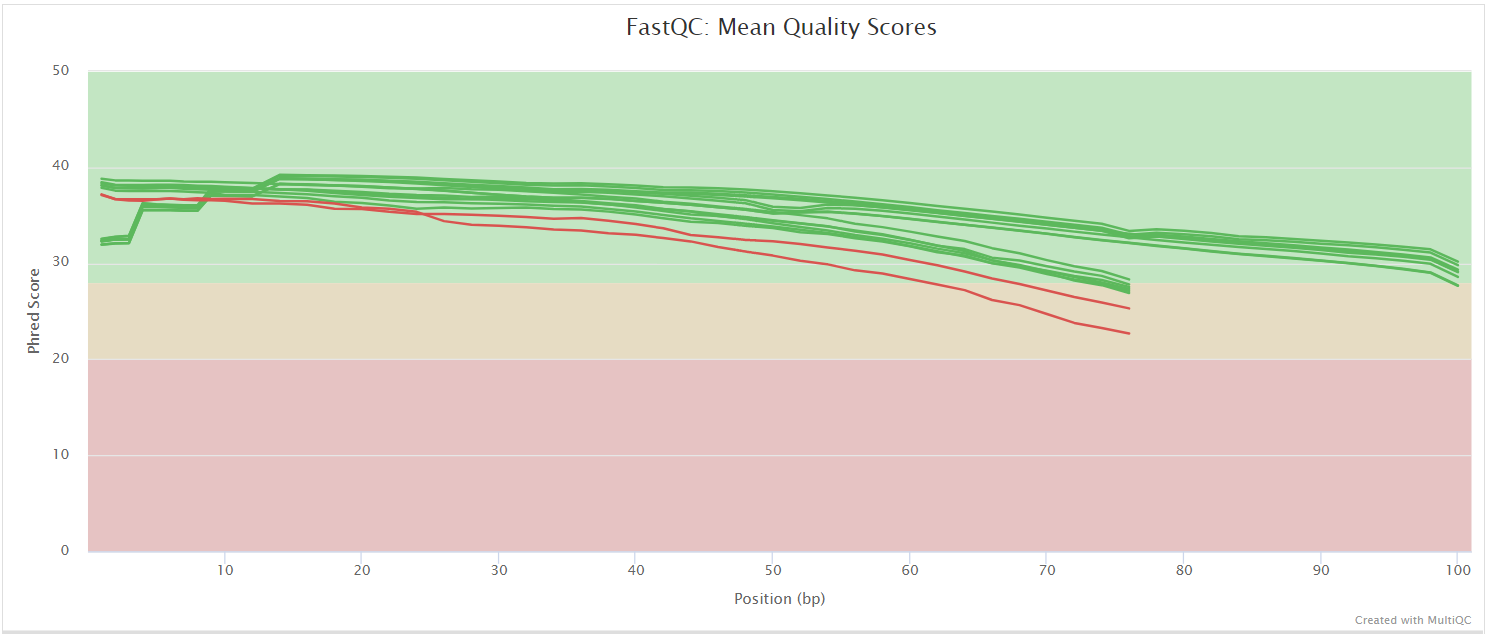
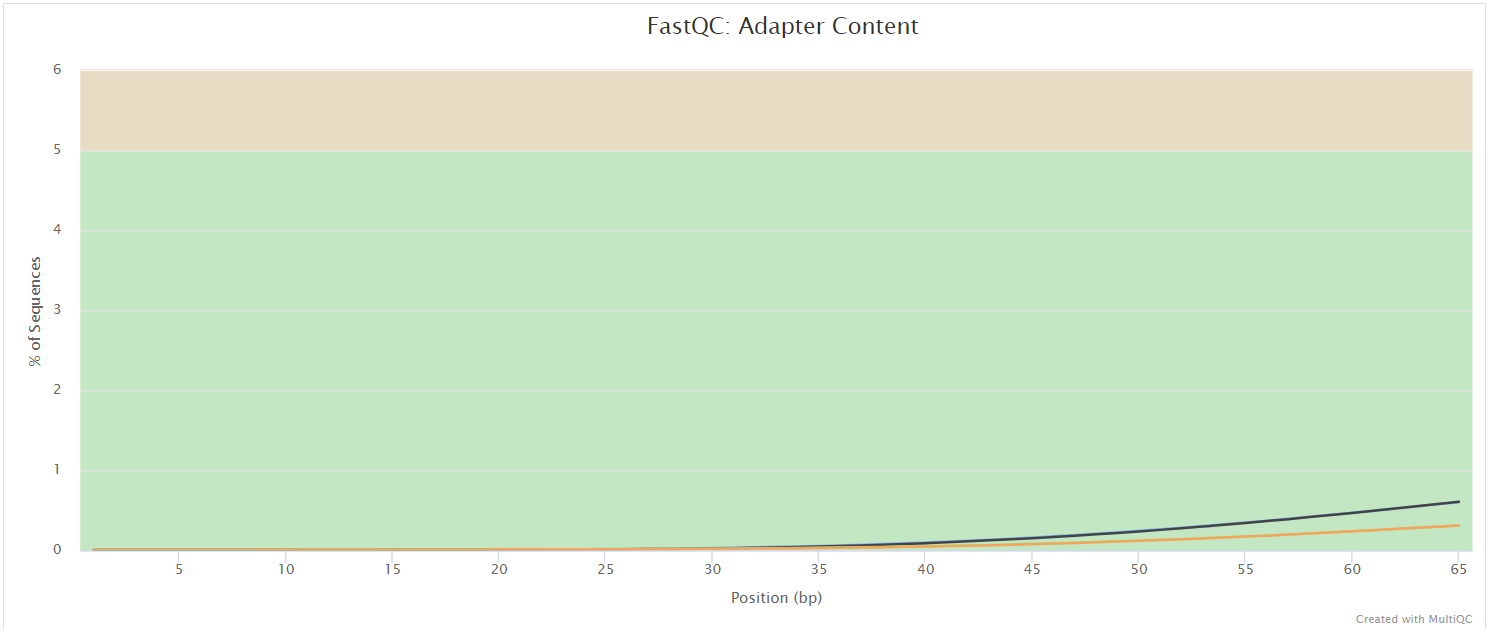
NB: The FastQC plots displayed in the MultiQC report shows untrimmed reads. They may contain adapter sequence and potentially regions with low quality.
MultiQC
Output files
multiqc/multiqc_report.html: a standalone HTML file that can be viewed in your web browser.multiqc_data/: directory containing parsed statistics from the different tools used in the pipeline.multiqc_plots/: directory containing static images from the report in various formats.
MultiQC is a visualization tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available in the report data directory.
Results generated by MultiQC collate pipeline QC from supported tools e.g. FastQC. The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability. For more information about how to use MultiQC reports, see http://multiqc.info.
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter’s are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv.
- Reports generated by Nextflow:
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.