nf-core/bactmap
A mapping-based pipeline for creating a phylogeny from bacterial whole genome sequences
0.9.1). The latest stable release is1.0.0.Introduction
This document describes the output produced by the pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
Reference indexingRead trimmingRead subsamplingRead mappingSort bam filesCall and filter variantsConvert filtered vcf to pseudogenomeCreate alignment from pseudogenomesRemove recombination (Optional)Remove non-informative positions- Construct phylogenetic tree (Optional)
- Pipeline information - Report metrics generated during the workflow execution
Reference Indexing
Output files
bwa/index.*.amb*.ann*.bwt*.pac*.sa
These files are generally not required except for in the mapping step
Read Trimming
Output files
fastp/*.htmlhtml reports of the trimming process that can be opened in any modern web browser*.jsontrimming report metrics in JSON computer readable formats
Read Subsampling
Output files
rasusa/*.fastq.gzsubsamples fastq files
Read Mapping
By default there are no output files since sorted bam files are produced in the next step
Sort Bam Files
Output files
samtools/*.bamsorted bam files*.bam.baibam file index*.bam.flagstatbam file metrics*.bam.idxstatsbam file metrics*.bam.statsbam file metrics
Call and Filter Variants
Output files
variants/*.vcf.gzfiltered vcf files containing variants
Convert Filtered VCF to Pseudogenome
Output files
pseudogenomes/*.faspseudogenome with a base at each position of the reference sequence
Create Alignment from Pseudogenomes
Only those pseudogenome fasta files that have a non-ACGT fraction less than the threshold specified will be included in the aligned_pseudogenomes.fas file. Those failing this will be reported in the low_quality_pseudogenomes.tsv file.
Output files
pseudogenomes/aligned_pseudogenomes.fasalignment of all sample pseudogenomes and the reference sequencelow_quality_pseudogenomes.tsva tab separated file of the samples that failed the non-ACGT base threshold
Remove Recombination
The file used for downstream tree building is aligned_pseudogenomes.filtered_polymorphic_sites.fasta. The other files are described in the gubbins documentation
Output files
gubbins/aligned_pseudogenomes.branch_base_reconstruction.emblaligned_pseudogenomes.filtered_polymorphic_sites.fastaaligned_pseudogenomes.filtered_polymorphic_sites.phylipaligned_pseudogenomes.final_tree.trealigned_pseudogenomes.node_labelled.final_tree.trealigned_pseudogenomes.per_branch_statistics.csvaligned_pseudogenomes.recombination_predictions.emblaligned_pseudogenomes.recombination_predictions.gffaligned_pseudogenomes.summary_of_snp_distribution.vcf
Remove Non-informative Positions
Output files
snpsites/constant.sites.txtA file with the number of constant sites for each basefiltered_alignment.fasAlignment with only informative positions (those positions that have at least one alternative variant base)
RapidNJ
Output files
rapidnj/rapidnj_phylogeny.treA newick tree built with RapidNJ
FastTree
Output files
fasttree/fasttree_phylogeny.treA newick tree built with FastTree
IQ-TREE
Output files
iqtree/*.treefileA ML tree built with IQ-TREE with support values for branches based on bootstrapping
RAxML-NG
Output files
iqtree/output.raxml.bestTreeA ML tree built with RAxML-NG selected as the best after running MLoutput.raxml.supportA ML tree built with RAxML-NG with support values for branches based on bootstrapping
MultiQC
Various quality statistics are compiled from the previous outputs using the MultiQC software:
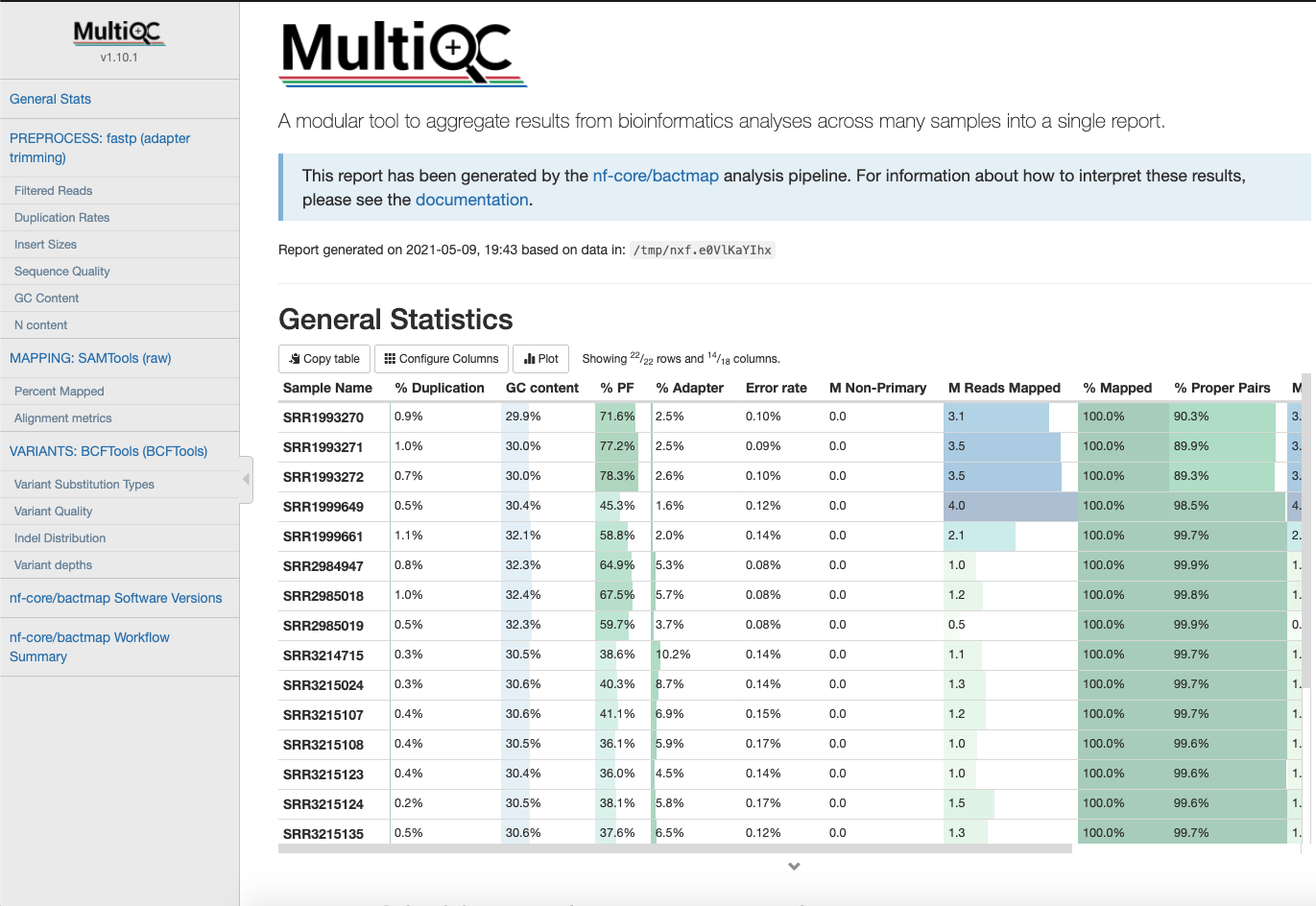
Overall Statistics
A compilation of statistics about read content, mapping and variants
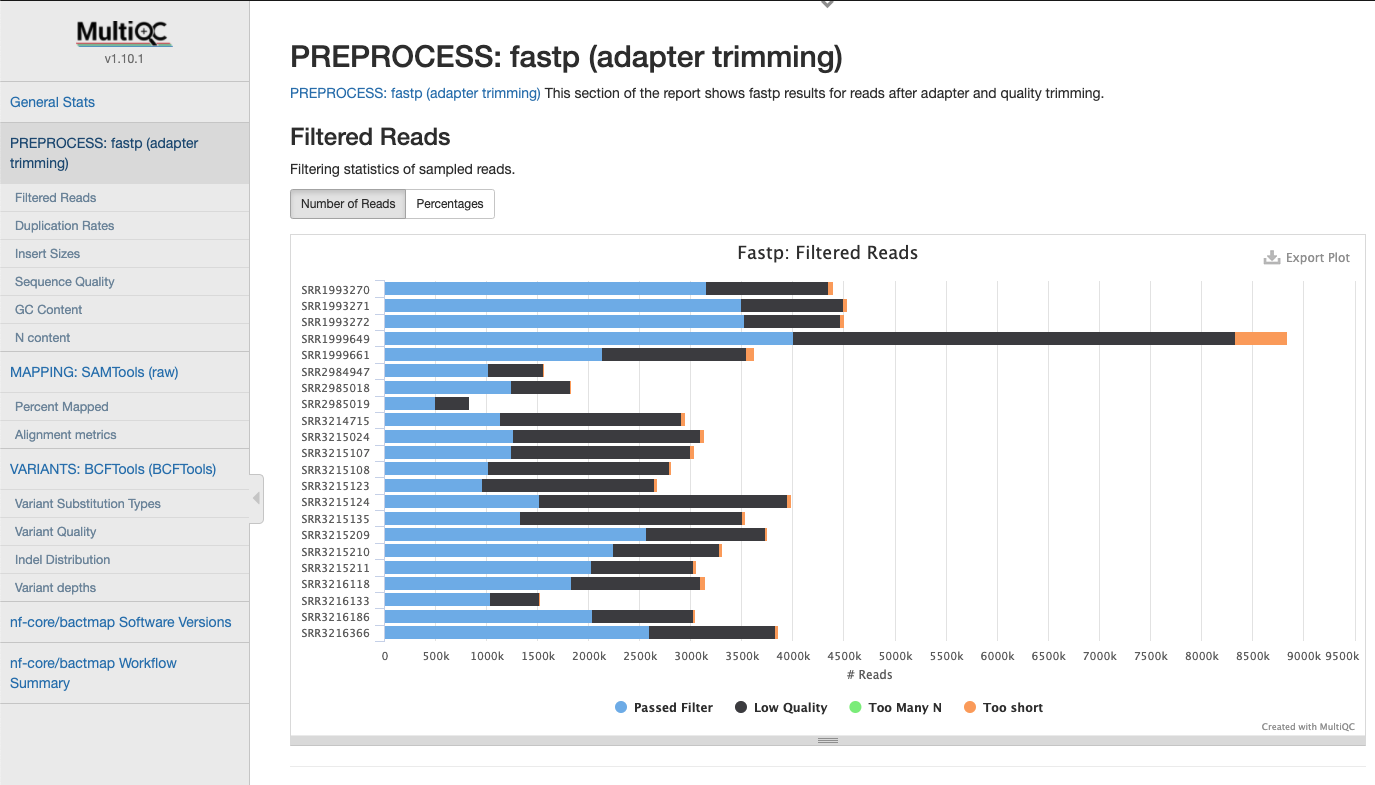
FastP Statistics
Statistics gathered when trimming reads
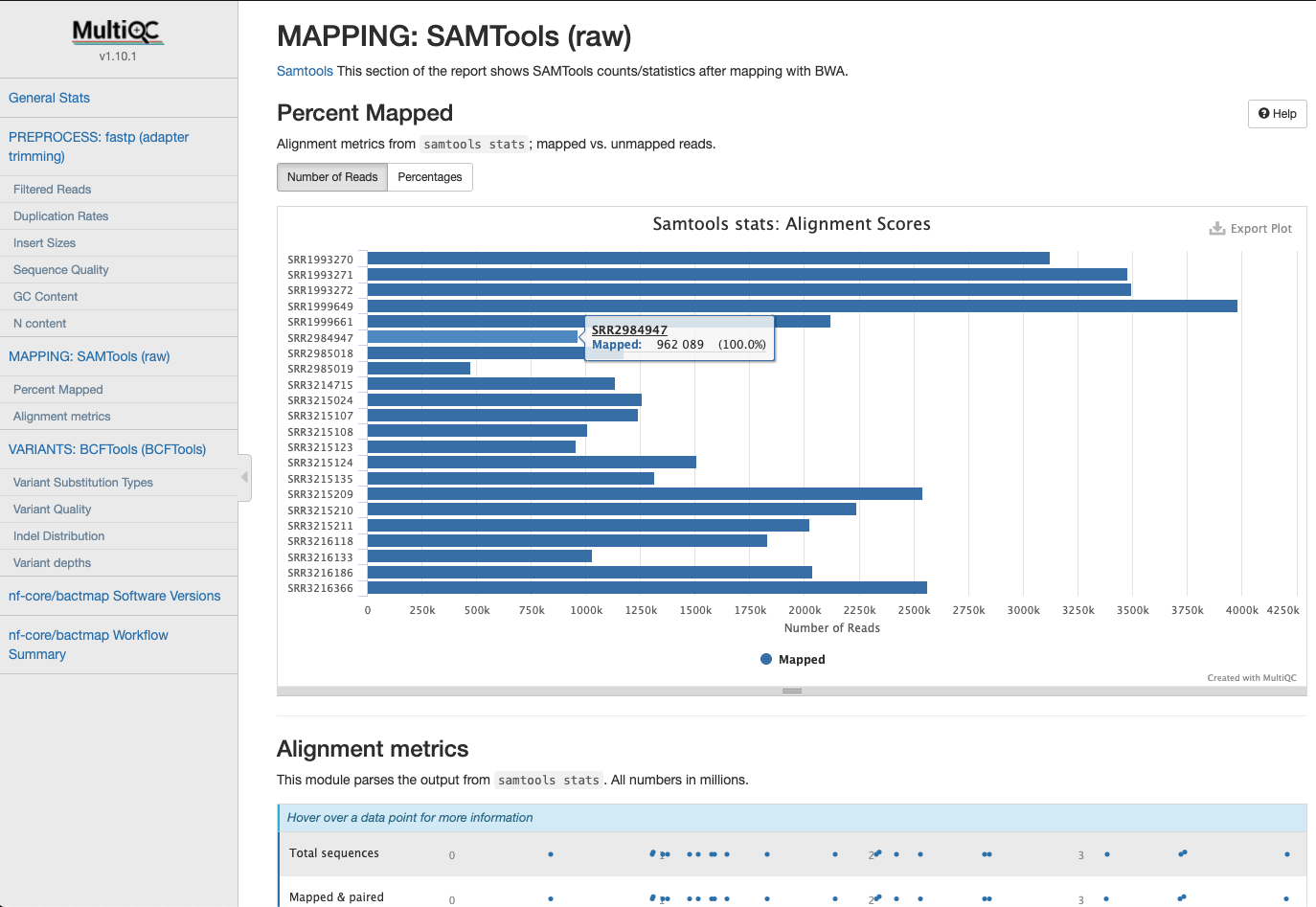
Mapping Statistics
Statistics gathered when mapping reads
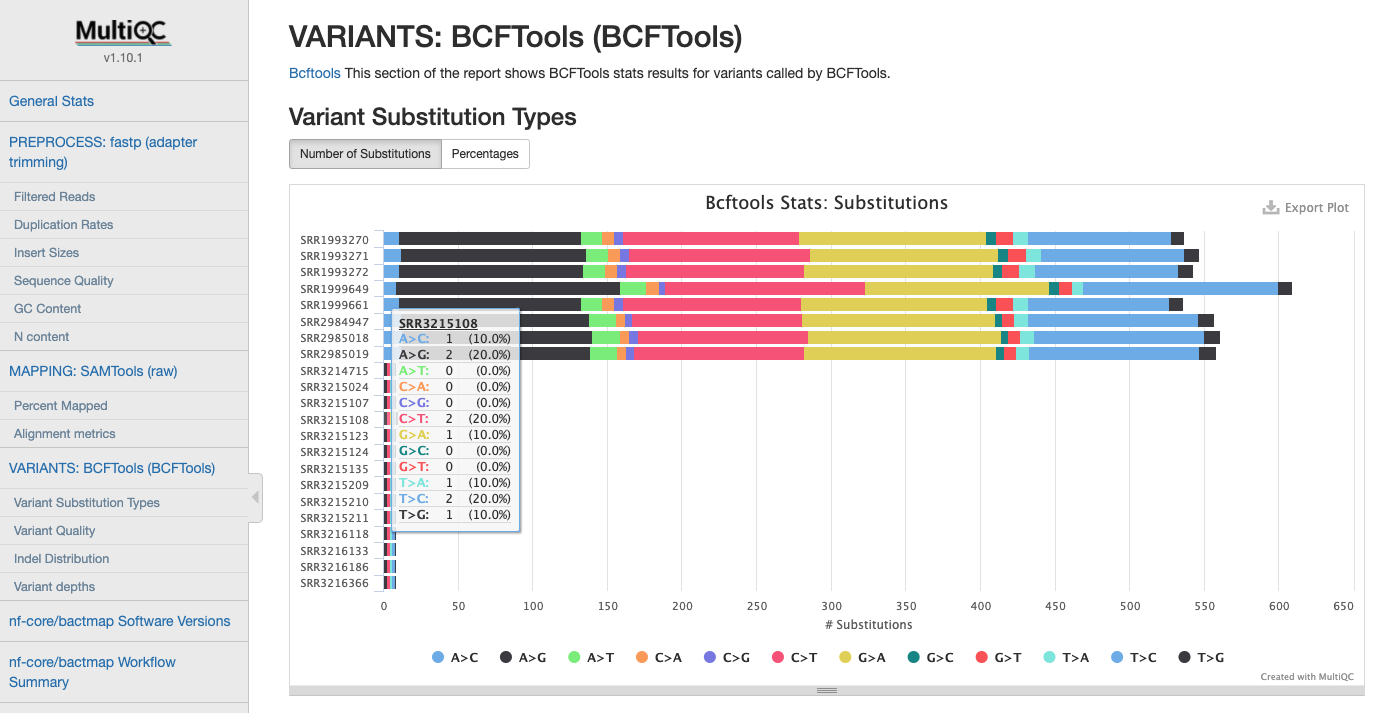
Varinat Statistics
Statistics gathered when calling variants after filtering
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.csv. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv.
- Reports generated by Nextflow:
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.