nf-core/datasync
nf-core/datasync is a system operation pipeline that provides several workflows for handling system operation / automation tasks
Introduction
nf-core/datasync is under active development and is not yet recommended for production data transfers. Validate its behaviour with representative data before relying on it.
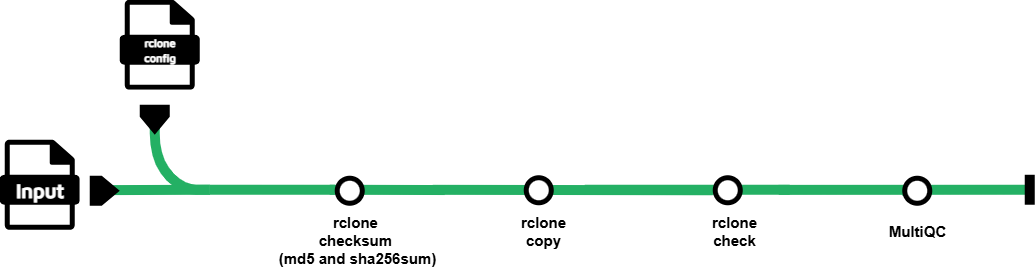
nf-core/datasync is a Nextflow pipeline for copying files and directories between storage locations and documenting their integrity. For every row in an input samplesheet, the pipeline:
- validates the source against a supplied MD5 and/or SHA-256 checksum manifest using
rclone checksum; - copies the source to the requested destination with
rclone copy; - compares the copied data with the source using
rclone check; and - produces detailed
rclonestatus files and a consolidated MultiQC report.
Sources and destinations may be local paths, HTTP(S) URLs, or rclone-supported remote storage such as Amazon S3, S3-compatible object storage, or Azure Blob Storage.
The current tested use case for this pipeline is transfer between S3 buckets.
Pass an rclone configuration with --rclone_config whenever a source or destination needs a configured remote, endpoint, or credentials. A single configuration file can contain separate named remotes for multiple providers; for non-S3 layouts, design and validate the provider-specific configuration using the upstream rclone documentation.
Quick start
If you are new to Nextflow and nf-core, see the nf-core environment setup guide. Nextflow 25.10.4 or later is required.
To explore the pipeline outputs before preparing your own data, run the bundled test profile with a container profile:
nextflow run nf-core/datasync \ -profile test,docker \ --outdir resultsThe test profile supplies a small samplesheet and rclone configuration automatically. It also enables --rclone_dry_run, so no files are actually transferred. This makes it useful for exploring the rclone/ output folders and multiqc/multiqc_report.html; remember that post-copy comparison reports describe whatever is already present at the destination because the dry run does not write transfer data.
To run the pipeline on your own data, create a samplesheet containing one transfer per row:
sample,input,output_path,checksum_md5,checksum_sharun_001,/data/run_001,s3://archive/runs,/data/manifests/run_001_md5.tsvreference,https://example.org/reference.fa,/data/references,,/data/manifests/reference_sha256.tsvThen launch the pipeline using:
nextflow run nf-core/datasync \ -r <VERSION> \ -profile docker \ --input samplesheet.csv \ --outdir results \ --rclone_config /path/to/rclone.conf--rclone_config is optional only when every source and destination is accessible without a configured rclone remote. See the rclone configuration section for the tested S3-to-S3 use case and guidance on adapting rclone configuration files for other providers. To preview copy operations without transferring data, add --rclone_dry_run; note that subsequent comparison reports will then describe the unchanged destination.
See the usage documentation for samplesheet rules, destination semantics, remote configuration, and reproducible execution. The complete generated parameter reference is available on the nf-core pipeline page.
Pipeline output
Results are written below --outdir. See the output documentation for file names and status-code interpretation.
Credits
nf-core/datasync was originally written by Alexander Peltzer.
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #datasync channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.