nf-core/deepmodeloptim
Stochastic Testing and Input Manipulation for Unbiased Learning Systems
Introduction
nf-core/deepmodeloptim augments your bio data towards an optimal task-specific training set.
Methods in deep learning are vastly equivalent (see neural scaling laws paper), most of the performance is driven by the training data.
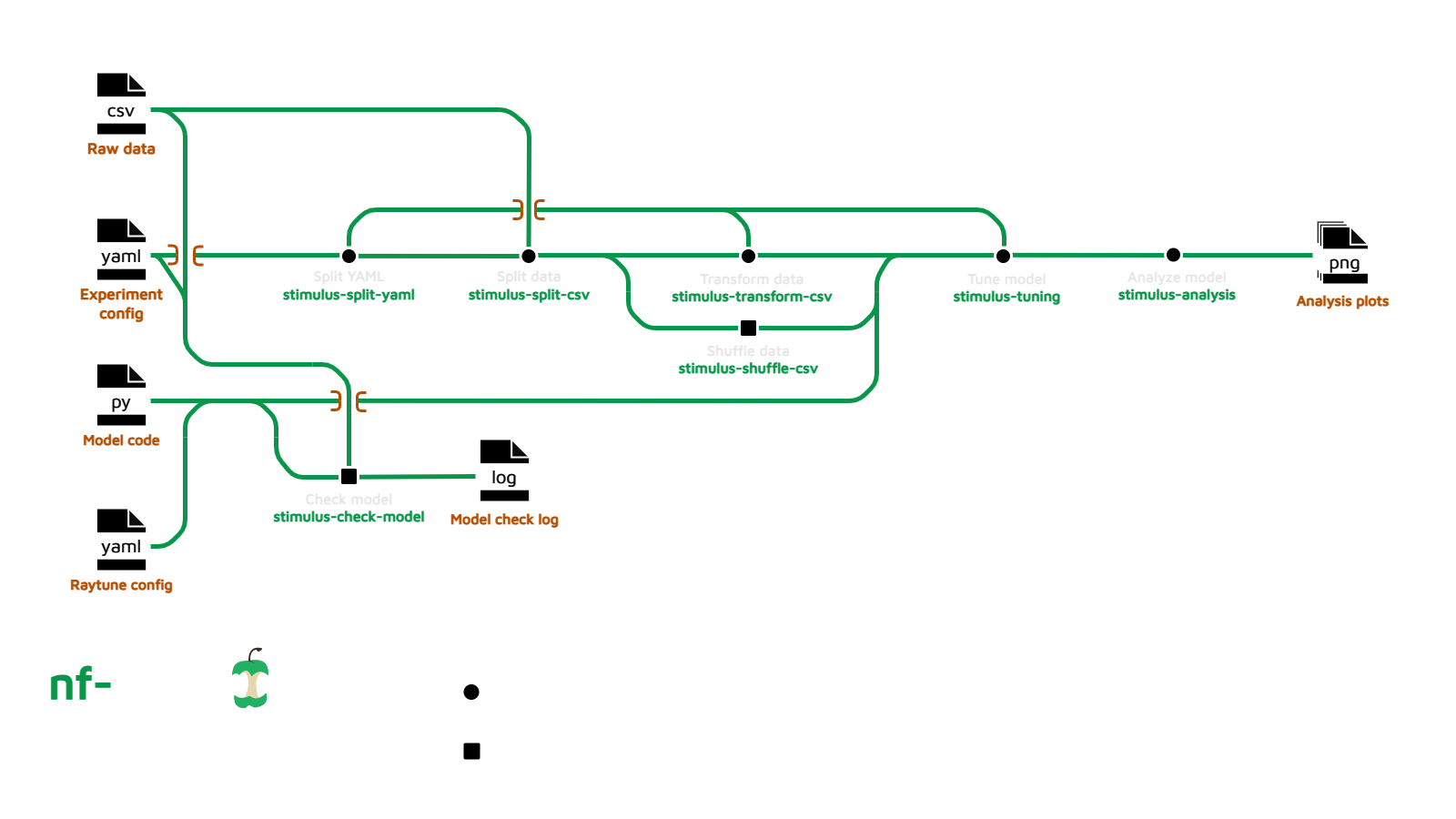
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Now, you can run the pipeline using:
nextflow run nf-core/deepmodeloptim \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Code requirements
Data
The data is provided as a csv where the header columns are in the following format : name:type:class
name is user given (note that it has an impact on experiment definition).
type is either “input”, “meta”, or “label”. “input” types are fed into the mode, “meta” types are registered but not transformed nor fed into the models and “label” is used as a training label.
class is a supported class of data for which encoding methods have been created, please raise an issue on github or contribute a PR if a class of your interest is not implemented
csv general example
| input1:input:input_type | input2:input:input_type | meta1:meta:meta_type | label1:label:label_type | label2:label:label_type |
|---|---|---|---|---|
| sample1 input1 | sample1 input2 | sample1 meta1 | sample1 label1 | sample1 label2 |
| sample2 input1 | sample2 input2 | sample2 meta1 | sample2 label1 | sample2 label2 |
| sample3 input1 | sample3 input2 | sample3 meta1 | sample3 label1 | sample3 label2 |
csv specific example
| mouse_dna:input:dna | mouse_rnaseq:label:float |
|---|---|
| ACTAGGCATGCTAGTCG | 0.53 |
| ACTGGGGCTAGTCGAA | 0.23 |
| GATGTTCTGATGCT | 0.98 |
Model
In STIMULUS, users input a .py file containing a model written in pytorch (see examples in bin/tests/models)
Said models should obey to minor standards:
- The model class you want to train should start with “Model”, there should be exactly one class starting with “Model”.
import torch
import torch.nn as nn
class SubClass(nn.Module):
"""
a subclass, this will be invisible to Stimulus
"""
class ModelClass(nn.Module):
"""
the PyTorch model to be trained by Stimulus, can use SubClass if needed
"""
class ModelAnotherClass(nn.Module):
"""
uh oh, this will return an error as there are two classes starting with Model
"""
- The model “forward” function should have input variables with the same names as the defined input names in the csv input file
import torch
import torch.nn as nn
class ModelClass(nn.Module):
"""
the PyTorch model to be trained by Stimulus
"""
def __init__():
# your model definition here
pass
def forward(self, mouse_dna):
output = model_layers(mouse_dna)
- The model should include a batch named function that takes as input a dictionary of input “x”, a dictionary of labels “y”, a Callable loss function and a callable optimizer.
In order to allow batch to take as input a Callable loss, we define an extra compute_loss function that parses the correct output to the correct loss class.
import torch
import torch.nn as nn
from typing import Callable, Optional, Tuple
class ModelClass(nn.Module):
"""
the PyTorch model to be trained by Stimulus
"""
def __init__():
# your model definition here
pass
def forward(self, mouse_dna):
output = model_layers(mouse_dna)
def compute_loss_mouse_rnaseq(self, output: torch.Tensor, mouse_rnaseq: torch.Tensor, loss_fn: Callable) -> torch.Tensor:
"""
Compute the loss.
`output` is the output tensor of the forward pass.
`mouse_rnaseq` is the target tensor -> label column name.
`loss_fn` is the loss function to be used.
IMPORTANT : the input variable "mouse_rnaseq" has the same name as the label defined in the csv above.
"""
return loss_fn(output, mouse_rnaseq)
def batch(self, x: dict, y: dict, loss_fn: Callable, optimizer: Optional[Callable] = None) -> Tuple[torch.Tensor, dict]:
"""
Perform one batch step.
`x` is a dictionary with the input tensors.
`y` is a dictionary with the target tensors.
`loss_fn` is the loss function to be used.
If `optimizer` is passed, it will perform the optimization step -> training step
Otherwise, only return the forward pass output and loss -> evaluation step
"""
output = self.forward(**x)
loss = self.compute_loss_mouse_rnaseq(output, **y, loss_fn=loss_fn)
if optimizer is not None:
optimizer.zero_grad()
loss.backward()
optimizer.step()
return loss, output
If you don’t want to optimize the loss function, the code above can be written in a simplified manner
import torch
import torch.nn as nn
from typing import Callable, Optional, Tuple
class ModelClass(nn.Module):
"""
the PyTorch model to be trained by Stimulus
"""
def __init__():
# your model definition here
pass
def forward(self, mouse_dna):
output = model_layers(mouse_dna)
def batch(self, x: dict, y: dict, optimizer: Optional[Callable] = None) -> Tuple[torch.Tensor, dict]:
"""
Perform one batch step.
`x` is a dictionary with the input tensors.
`y` is a dictionary with the target tensors.
`loss_fn` is the loss function to be used.
If `optimizer` is passed, it will perform the optimization step -> training step
Otherwise, only return the forward pass output and loss -> evaluation step
"""
output = self.forward(**x)
loss = nn.MSELoss(output, y['mouse_rnaseq'])
if optimizer is not None:
optimizer.zero_grad()
loss.backward()
optimizer.step()
return loss, output
Model parameter search design
Experiment design
The file in which all information about how to handle the data before tuning is called an experiment_config. This file in .json format for now but it will be soon moved to .yaml. So this section could vary in the future.
The experiment_config is a mandatory input for the pipeline and can be passed with the flag --exp_conf followed by the PATH of the file you want to use. Two examples of experiment_config can be found in the examples directory.
Experiment config content description.
Credits
nf-core/deepmodeloptim was originally written by Mathys Grapotte (@mathysgrapotte).
We would like to thank to all the contributors for their extensive assistance in the development of this pipeline, who include (but not limited to):
- Alessio Vignoli (@alessiovignoli)
- Suzanne Jin (@suzannejin)
- Luisa Santus (@luisas)
- Jose Espinosa (@JoseEspinosa)
- Evan Floden (@evanfloden)
- Igor Trujnara (@itrujnara)
Special thanks for the artistic work on the logo to Maxime (@maxulysse), Suzanne (@suzannejin), Mathys (@mathysgrapotte) and, not surprisingly, ChatGPT.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #deepmodeloptim channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.