nf-core/demultiplex
Demultiplexing pipeline for sequencing data
Introduction
This document describes the output produced by the pipeline. Most of the plots are taken from the MultiQC report, which summarises results at the end of the pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
- bcl-convert - converting bcl files to fastq, and demultiplexing (CONDITIONAL)
- bases2fastq - converting bases files to fastq, and demultiplexing (CONDITIONAL)
- bcl2fastq - converting bcl files to fastq, and demultiplexing (CONDITIONAL)
- sgdemux - demultiplexing bgzipped fastq files produced by Singular Genomics (CONDITIONAL)
- fqtk - demultiplexing fastq files (CONDITIONAL)
- mkfastq - converting bcl files to fastq, and demultiplexing for single-cell sequencing data (CONDITIONAL)
- mgikit - Demultiplex fastq files generated by MGI sequencers using mgikit (CONDITIONAL).
- checkqc - (optional) Check quality criteria after demultiplexing (bcl2fastq only)
- fastp - Adapter and quality trimming
- Falco - Raw read QC
- md5sum - Creates an MD5 (128-bit) checksum of every fastq.
- kraken2 - Kraken2 is a taxonomic sequence classifier that assigns taxonomic labels to sequence reads.
- samplesheet - Samplesheet generation for downstream nf-core pipelines.
- MultiQC - aggregate report, describing results of the whole pipeline
bcl-convert
bcl-convert Conversion Software both demultiplexes data and converts BCL files generated by Illumina sequencing systems to standard FASTQ file formats for downstream analysis.
Output files
<flowcell_id>/sample.fastq.gzor<flowcell_id>/L00<meta.lane>/sample.fastq.gz- Untrimmed raw fastq files
bases2fastq
bases2fastq demultiplexes sequencing data and converts base calls generated by the Element AVITI™ System into FASTQ files for secondary analysis with FASTQ-compatible software.
Output files
| File | Directory | Description |
|---|---|---|
bases2fastq.log |
info | Log file that records software events |
| HTML QC report | Root | Interactive report on run performance and quality |
| FASTQ | Samples/{sample} | The primary output of Bases2Fastq |
Metrics.csv |
Root | Statistics for each lane: mismatch rates, percent assigned, and per-sample yield |
RunManifest.csv |
Root | The AOS- or user-created run manifest |
RunManifestErrors.json |
info | A record of errors in the run manifest |
RunParameters.json |
Root | A copy of the original run parameters file |
RunStats.json |
Root | Information on run performance |
| Sample statistics | Samples/{sample} | Information on the performance of each sample in the run |
UnassignedSequences.csv |
Root | The most frequent unassigned index sequences with approximate counts* |
bcl2fastq
sgdemux
sgdemux demultiplexes sequencing data generated on Singular Genomics sequencing instruments.
Output files
| File | Directory | Description |
|---|---|---|
| Execution logs | Log files for the nextflow workflow | |
software_versions.yml |
Log file with software versions | |
| FASTQ | Demultiplexed fastq.gz files | |
metrics.csv |
Summary stats across run: control_reads_omitted, failing_reads_omitted, and total_templates | |
most_frequent_unmatched.tsv |
Counts for the most prevalent unmatched barcode: barcode, count | |
per_project_metrics.tsv |
Summary metrics per project | |
per_sample_metrics.tsv |
Summary metrics per sample | |
sample_barcode_hop_metrics.tsv |
Unexpected barcode combinations for dual indexed reads, else empty file | |
| Fastqc summary stats | Per base quality summary, for each demultiplexed fastq file | |
| Fastq summary html | Interactive html link for fastqc summary stats | |
| Md5Sum | Md5Sums for each demultiplexed fastq file |
checkqc
checkqc - (optional) CheckQC is a program designed to check a set of quality criteria against an Illumina runfolder. Available for outputs from bcl2fastq only. The program will summarize the type of run it has identified and output any warnings and/or errors in finds. The CheckQC module in demultiplex will output the summary of the QC, along with its warnings and errors in the checkqc_report.json. In addition, a checkqc_log.txt will contain the log of the program for inspection. If the run directory misses some input files, it will return a non-zero exit status and also the information in the checkqc_log.txt.
Output files
checkqc/checkqc_report.json- QC report of bcl2fastq run
fqtk
fqtk A toolkit for working with FASTQ files, written in Rust.
Output files
| File | Directory | Description |
|---|---|---|
| Execution logs | Log files for the nextflow workflow | |
software_versions.yml |
Log file with software versions | |
| FASTQ | Demultiplexed fastq.gz files | |
demux-metrics.txt |
Summary stats across run | |
unmatched_<1/2>.fastq.gz |
Unmatched R1 and R2 records | |
| Fastqc summary stats | Per base quality summary, for each demultiplexed fastq file | |
| Fastq summary html | Interactive html link for fastqc summary stats | |
| Md5Sum | Md5Sums for each demultiplexed fastq file |
mkfastq
mkfastq A tool for converting BCL files to FASTQ and demultiplexing for single-cell sequencing data.
Output files
| File | Directory | Description |
|---|---|---|
| FASTQ | Demultiplexed fastq.gz files | |
*.fastp.html |
HTML report for fastp | |
*.fastp.json |
JSON report for fastp | |
*_summary.txt |
Summary statistics for the sequencing run | |
| Fastqc summary stats | Per base quality summary, for each demultiplexed FASTQ file | |
| Fastqc summary html | Interactive html link for fastqc summary stats | |
| Md5Sum | Md5Sums for each demultiplexed FASTQ file |
mgikit
mgikit demultiplexes fastq files generated by MGI sequencers (CONDITIONAL).
fastp
Output files
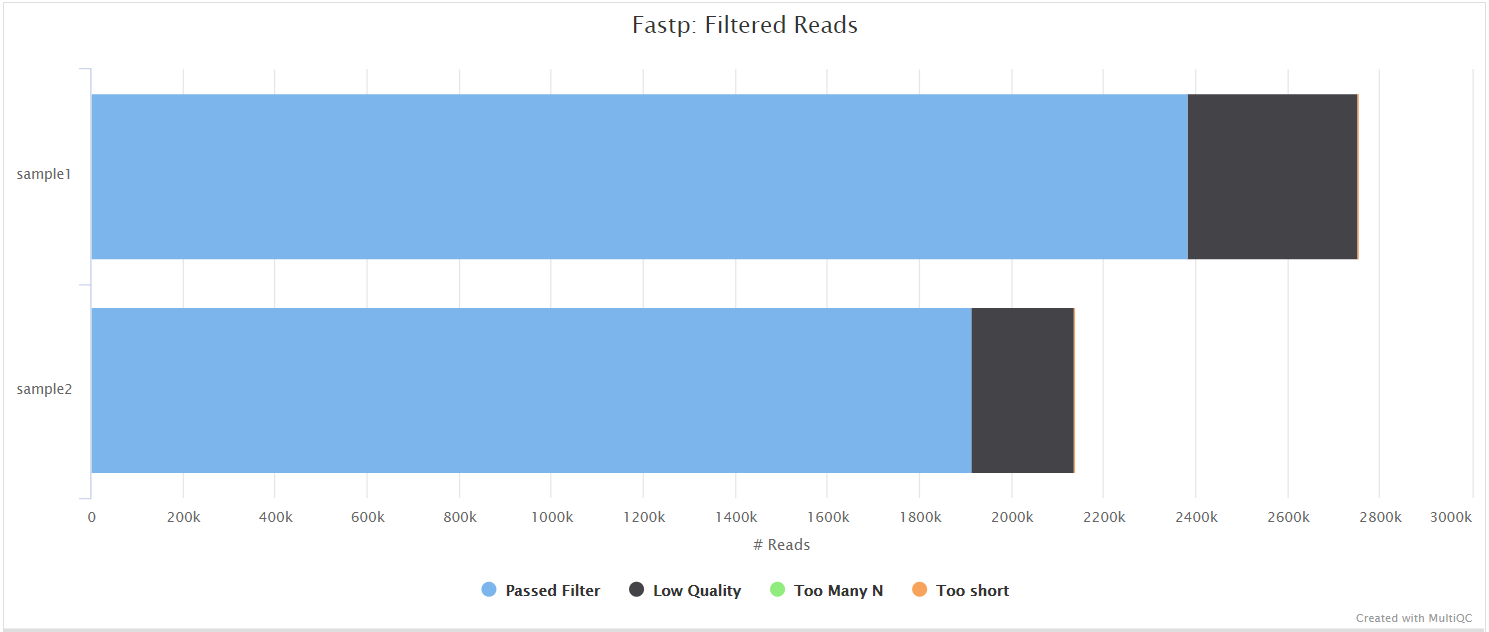
<flowcell_id>/or<flowcell_id>/L00<meta.lane>/*.fastp.html: Trimming report in html format.*.fastp.json: Trimming report in json format.*.fastp.log: Trimming log file.
fastp is a tool designed to provide fast, all-in-one preprocessing for FastQ files. It has been developed in C++ with multithreading support to achieve higher performance. fastp is used in this pipeline for standard adapter trimming and quality filtering.
Falco
Output files
<flowcell_id>/or<flowcell_id>/L00<meta.lane>/*_fastqc.html: FastQC report containing quality metrics.*_fastqc.txt: Txt containing the FastQC report, tab-delimited data file.*_summary.txt: Txt containing the summary metrics.
NB: The FastQC plots in this directory are generated relative to the raw, demultiplexed reads. They may contain adapter sequence and regions of low quality.
Falco gives general quality metrics about your sequenced reads. It provides information about the quality score distribution across your reads, per base sequence content (%A/T/G/C), adapter contamination and overrepresented sequences.



The FastQC plots displayed in the MultiQC report shows untrimmed reads. They may contain adapter sequence and potentially regions with low quality.
md5sum
Output files
<flowcell_id>/or<flowcell_id>/L00<meta.lane>/*.fastq.gz.md5: MD5 checksum of fastq
Creates an MD5 (128-bit) checksum of every fastq.
kraken2
Output files
<flowcell_id>/or<flowcell_id>/L00<meta.lane>/*.*report.txt: A Kraken2 report that summarises the fraction abundance, taxonomic ID, number of Kmers, taxonomic path of all the hits in the Kraken2 run for a given sample.*.classified.fastq.gz: FASTQ file containing all reads that had a hit against a reference in the database for a given sample.*.unclassified.fastq.gz: FASTQ file containing all reads that did not have a hit in the database for a given sample.*.classifiedreads.txt: A list of read IDs and the hits each read had against each database for a given sample.
Kraken is a taxonomic sequence classifier that assigns taxonomic labels to DNA sequences. Kraken examines the k-mers within a query sequence and uses the information within those k-mers to query a database. That database maps -mers to the lowest common ancestor (LCA) of all genomes known to contain a given k-mer.
Downstream pipeline samplesheets
Output files
<outputdir>/samplesheet/*.csv: Samplesheets with the generated FASTQ files formatted according to the selected downstream nf-core pipeline. Currently, samplesheets are created for the pipelines atacseq, methylseq, rnaseq, sarek, taxprofiler.
Adapter sequence removal from samplesheet
Output files
<flowcell_id>.lane<meta.lane>_no_adapters.csv
This is done by a custom function in the workflow, not by a module. Creates an updated samplesheet from the input by removing the adapter sequence within the “[Settings]” section. If the samplesheet doesn’t require any adapter removal, it will still be published in the specified output folder.
MultiQC
Output files
multiqc/multiqc_report.html: a standalone HTML file that can be viewed in your web browser.multiqc_data/: directory containing parsed statistics from the different tools used in the pipeline.multiqc_plots/: directory containing static images from the report in various formats.
MultiQC is a visualization tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available in the report data directory.
Results generated by MultiQC collate pipeline QC from supported tools e.g. FastQC. The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability. For more information about how to use MultiQC reports, see http://multiqc.info.
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter’s are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv. - Parameters used by the pipeline run:
params.json.
- Reports generated by Nextflow:
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.