nf-core/dualrnaseq
Analysis of Dual RNA-seq data - an experimental method for interrogating host-pathogen interactions through simultaneous RNA-seq.
22.10.6.
Learn more.
![]()
![]()
![]()
![]()

Dual RNA-seq pipeline
nf-core/dualrnaseq is a bioinformatics pipeline built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible.
Introduction
nf-core/dualrnaseq is specifically used for the analysis of Dual RNA-seq data, interrogating host-pathogen interactions through simultaneous RNA-seq.
This pipeline has been initially tested with eukaryotic host’s including Human and Mouse, and pathogens including Salmonella enterica, Orientia tsutsugamushi, Streptococcus penumoniae, Escherichia coli and Mycobacterium leprae. The workflow should work with any eukaryotic and bacterial organisms with an available reference genome and annotation.
Method
The workflow merges host and pathogen genome annotations taking into account differences in annotation conventions, then processes raw data from FastQ inputs (FastQC, BBDuk), quantifies gene expression (STAR and HTSeq; STAR, Salmon and tximport; or Salmon in quasimapping mode and tximport), and summarises the results (MultiQC), as well as generating a number of custom summary plots and separate results tables for the pathogen and host. See the output documentation for more details.
Workflow
The workflow diagram below gives a simplified visual overview of how dualrnaseq has been designed.
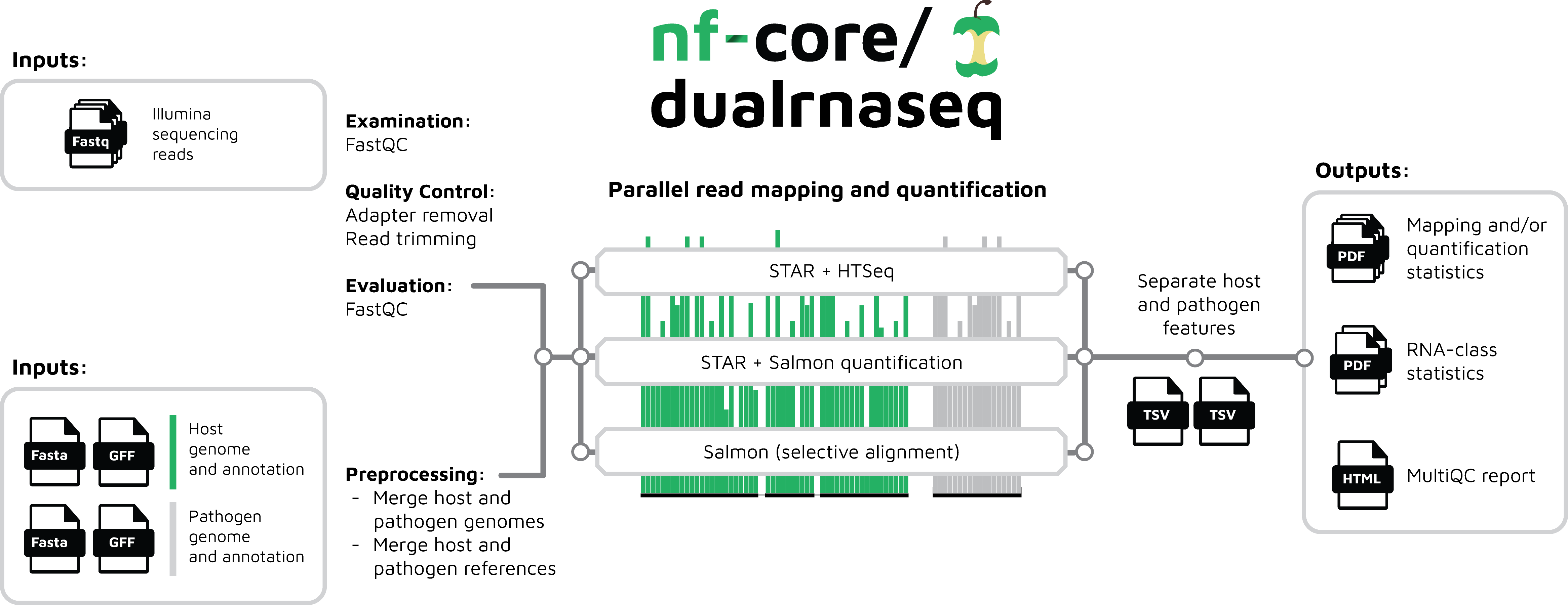
Documentation
The nf-core/dualrnaseq pipeline comes with documentation about the pipeline, found in the docs/ directory:
- Installation
- Pipeline configuration
- Running the pipeline
- Output and how to interpret the results
- Troubleshooting
Credits
nf-core/dualrnaseq was coded and written by Bozena Mika-Gospodorz and Regan Hayward.
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #dualrnaseq channel (you can join with this invite).
Citations
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x. ReadCube: Full Access Link
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.