nf-core/eager
A fully reproducible and state-of-the-art ancient DNA analysis pipeline
22.10.6.Learn more.Introduction
The output of nf-core/eager primarily consists of the following main components: output alignment files (e.g. VCF, BAM or FASTQ files), and summary statistics of the whole run presented in a MultiQC report. Intermediate files and module-specific statistics files are also retained depending on your particular run configuration.
Directory Structure
The default directory structure of nf-core/eager is as follows
results/├── MultiQC/├── <MODULE_1>/├── <MODULE_2>/├── <MODULE_3>/├── pipeline_info/└── reference_genome/work/- The parent directory
<RUN_OUTPUT_DIRECTORY>is the parent directory of the run, either the directory the pipeline was run from or as specified by the--outdirflag. The default name of the output directory (unless otherwise specified) will be./results/.
Primary Output Directories
These directories are the ones you will use on a day-to-day basis and are those which you should familiarise yourself with.
- The
MultiQCdirectory is the most important directory and contains the main summary report of the run in HTML format, which can be viewed in a web-browser of your choice. The sub-directory contains the MultiQC collected data used to build the HTML report. The Report allows you to get an overview of the sequencing and mapping quality as well as aDNA metrics (see the MultiQC Report section for more detail). - A
<MODULE>directory contains the (cleaned-up) output from a particular software module. This is the second most important set of directories. This contains output files such as FASTQ, BAM, statistics, and/or plot files of a specific module (see the Output Files section for more detail). The latter two are only needed when you need finer detail about that particular part of the pipeline.
Secondary Output Directories
These are less important directories which are used less often, normally in the context of bug-reporting.
pipeline_info/: Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.csv. - Documentation for interpretation of results in HTML format:
results_description.html.
- Reports generated by Nextflow:
reference_genome/contains either text files describing the location of specified reference genomes, and if not already supplied when running the pipeline, auxiliary indexing files. This is often useful when re-running other samples using the same reference genome, but is otherwise often not important.- The
work/directory contains all thenextflowprocessing directories. This is wherenextflowactually does all the work, but in an efficient programmatic procedure that is not intuitive to human-readers. Due to this, the directory is often not important to a user as all the useful output files are linked to the module directories (see above). Otherwise, this directory maybe useful when a bug-reporting.
⚠️ Note that
work/will be created wherever you are running thenextflow runcommand from, unless you specify the location with-w, i.e. it will not by default be inoutdir!.
MultiQC Report
In this section we will run through the output of each default module as reported in a MultiQC output. This can be viewed by opening the HTML file in your <RUN_OUTPUT_DIRECTORY>/MultiQC/ directory in a web browser. The section will also provide some basic tips on how to interpret the plots and values, although we highly recommend reading the READMEs or original papers of the tools used in the pipeline. A list of references can be seen on the nf-core/eager github repository
For more information about how to use MultiQC reports, see http://multiqc.info
General Stats Table
Background
This is the main summary table produced by MultiQC that the report begins with. This section of the report is generated by MultiQC itself rather than stats produced by a specific module. It shows whatever each module considers to be as the ‘most important’ values to be displayed — however the nf-core/eager version has been somewhat customised to make it as close to the EAGER (v1) ReportTable format as possible, with some opinionated tweaks.
Table
This table will report values per-file, library, or sample statistics depending on which stage along the pipeline you have gone through. MultiQC will try and collapse the rows as far as possible, if the log files have the same name. However, separate libraries will be displayed separately, for example when using DamageProfiler with the using TSV input and merging of samples is performed (which would be reported at the qualimap level). If you’re only interested in a single part of the results (e.g. qualimap) you can use the Configure Columns to remove columns and the corresponding rows will be not displayed, resulting in a more compact table.
Each column name is supplied by the module, so you may see similar column names. When unsure, hovering over the column name will allow you see which module it is derived from.
The possible columns displayed by default are as follows (note you may see additional columns depending on what other modules you activate):
- Sample Name This is the log file name without file suffix(s). This will depend on the module outputs.
- Nr. Input Reads This is from Pre-AdapterRemoval FastQC. Represents the number of raw reads in your untrimmed and (paired end) unmerged FASTQ file. Each row should be approximately equal to the number of reads you requested to be sequenced, divided by the number of FASTQ files you received for that library.
- Length Input Reads This is from Pre-AdapterRemoval FastQC. This is the average read length in your untrimmed and (paired end) unmerged FASTQ file and should represent the number of cycles of your sequencing chemistry.
- % GC Input Reads This is from Pre-AdapterRemoval FastQC. This is the average GC content in percent of all the reads in your untrimmed and (paired end) unmerged FASTQ file.
- GC content This is from FastP. This is the average GC of all reads in your untrimmed and unmerged FASTSQ file after poly-G tail trimming. If you have lots of tails, this value should drop from the pre-AdapterRemoval FastQC %GC column.
- % Trimmed This is from AdapterRemoval. It is the percentage of reads which had an adapter sequence removed from the end of the read.
- Nr. Processed Reads This is from Post-AdapterRemoval FastQC. Represents the number of preprocessed reads in your adapter trimmed (paired end) merged FASTQ file. The loss between this number and the Pre-AdapterRemoval FastQC can give you an idea of the quality of trimming and merging.
- % GC Processed Reads This is from Post-AdapterRemoval FastQC. Represents the average GC of all preprocessed reads in your adapter trimmed (paired end) merged FASTQ file.
- Length Processed Reads This is from post-AdapterRemoval FastQC. This is the average read length in your trimmed and (paired end) merged FASTQ file and should represent the ‘realistic’ average lengths of your DNA molecules
- % Aligned This is from bowtie2. It reports the percentage of input reads that mapped to your reference genome. This number will be likely similar to Endogenous DNA % (see below).
- % Metagenomic Mappability This is from MALT. It reports the percentage of the off-target reads (from mapping), that could map to your MALT metagenomic database. This can often be low for aDNA due to short reads and database bias.
- % Unclassified This is from Kraken. It reports the percentage of reads that could not be aligned and taxonomically assigned against your Kraken metagenomic database. This can often be high for aDNA due to short reads and database bias.
- Nr. Reads Into Mapping This is from Samtools. This is the raw number of preprocessed reads that went into mapping.
- Nr. Mapped Reads This is from Samtools. This is the raw number of preprocessed reads mapped to your reference genome prior map quality filtering.
- Endogenous DNA (%) This is from the endorS.py tool. It displays a percentage of mapped reads over total reads that went into mapped (i.e. the percentage DNA content of the library that matches the reference). Assuming a perfect ancient sample with no modern contamination, this would be the amount of true ancient DNA in the sample. However this value most likely include contamination and will not entirely be the true ‘endogenous’ content.
- Nr. Mapped Reads Post-Filter This is from Samtools. This is the raw number of preprocessed reads mapped to your reference genome after map quality filtering (note the column name does not distinguish itself from prior-map quality filtering, but the post-filter column is always second)
- Endogenous DNA Post-Filter (%) This is from the endorS.py tool. It displays a percentage of mapped reads after BAM filtering (i.e. for mapping quality and/or bam-level length filtering) over total reads that went into mapped (i.e. the percentage DNA content of the library that matches the reference). This column will only be displayed if BAM filtering is turned on and is based on the original mapping for total reads, and mapped reads as calculated from the post-filtering BAM.
- ClusterFactor This is from DeDup only. This is a value representing how many duplicates in the library exist for each unique read. This ratio is calculated as
reads_before_deduplication / reads_after_deduplication. Can be converted to %Dups by calculating1 - (1 / CF). A cluster factor close to one indicates a highly complex library and could be sequenced further. Generally with a value of more than 2 you will not be gaining much more information by sequencing deeper. - % Dup. Mapped Reads This is from Picard’s markDuplicates only. It represents the percentage of reads in your library that were exact duplicates of other reads in your library. The lower the better, as high duplication rate means lots of sequencing of the same information (and therefore is not time or cost effective).
- X Prime Y>Z N base These columns are from DamageProfiler or mapDamage. The prime numbers represent which end of the reads the damage is referring to. The Y>Z is the type of substitution (C>T is the true damage, G>A is the complementary). You should see for no- and half-UDG treatment a decrease in frequency from the 1st to 2nd base.
- Mean Length Mapped Reads This is from DamageProfiler. This is the mean length of all de-duplicated mapped reads. Ancient DNA normally will have a mean between 30-75, however this can vary.
- Median Length Mapped Reads This is from DamageProfiler. This is the median length of all de-duplicated mapped reads. Ancient DNA normally will have a mean between 30-75, however this can vary.
- Nr. Dedup. Mapped Reads This is from Qualimap. This is the total number of deduplicated reads that mapped to your reference genome. This is the best number to report for final mapped reads in final publications.
- Mean/Median Coverage This is from Qualimap. This is the mean/median number of times a base on your reference genome was covered by a read (i.e. depth coverage). This average includes bases with 0 reads covering that position.
- >= 1X to >= 5X These are from Qualimap. This is the percentage of the genome covered at that particular depth coverage.
- % GC Dedup. Mapped Reads This is the mean GC content in percent of all mapped reads post-deduplication. This should normally be close to the GC content of your reference genome.
- MT to Nuclear Ratio This from MTtoNucRatio. This reports the number of reads aligned to a mitochondrial entry in your reference FASTA to all other entries. This will typically be high but will vary depending on tissue type.
- SexDet Rate X Chr This is from Sex.DetERRmine. This is the relative depth of coverage on the X-chromosome.
- SexDet Rate Y Chr This is from Sex.DetERRmine. This is the relative depth of coverage on the Y-chromosome.
- #SNPs Covered This is from eigenstrat_snp_coverage. The number of called SNPs after genotyping with pileupcaller.
- #SNPs Total This is from eigenstrat_snp_coverage. The maximum number of covered SNPs, i.e. the number of SNPs in the .snp file provided to pileupcaller with
--pileupcaller_snpfile. - Number of SNPs This is from ANGSD. The number of SNPs left after removing sites with no data in a 5 base pair surrounding region.
- Contamination Estimate (Method1_ML) This is from the nuclear contamination function of ANGSD. The Maximum Likelihood contamination estimate according to Method 1. The estimates using Method of Moments and/or those based on Method 2 can be unhidden through the “Configure Columns” button.
- Estimate Error (Method1_ML) This is from ANGSD. The standard error of the Method1 Maximum likelihood estimate. The errors associated with Method of Moments and/or Method2 estimates can be unhidden through the “Configure Columns” button.
- % Hets This is from MultiVCFAnalyzer. This reports the number of SNPs on an assumed haploid organism that have two possible alleles. A high percentage may indicate cross-mapping from a related species.
For other non-default columns (activated under ‘Configure Columns’), hover over the column name for further descriptions.
FastQC
Background
FastQC gives general quality metrics about your raw reads. It provides information about the quality score distribution across your reads, the per base sequence content (%T/A/G/C) as sequenced. You also get information about adapter contamination and other overrepresented sequences.
You will receive output for each supplied FASTQ file.
When dealing with ancient DNA data the MultiQC plots for FastQC will often show lots of ‘warning’ or ‘failed’ samples. You generally can discard this sort of information as we are dealing with very degraded and metagenomic samples which have artefacts that violate the FastQC ‘quality definitions’, while still being valid data for aDNA researchers. Instead you will normally be looking for ‘global’ patterns across all samples of a sequencing run to check for library construction or sequencing failures. Decision on whether a individual sample has ‘failed’ or not should be made by the user after checking all the plots themselves (e.g. if the sample is consistently an outlier to all others in the run).
FastQC gives general quality metrics about your sequenced reads. It provides information about the quality score distribution across your reads, per base sequence content (%A/T/G/C), adapter contamination and overrepresented sequences.
For further reading and documentation see the FastQC help pages.
NB: The FastQC (pre-Trimming) plots displayed in the MultiQC report shows untrimmed reads. They may contain adapter sequence and potentially regions with low quality. To see how your reads look after trimming, look at the FastQC reports in the FastQC (post-Trimming) section. You should expect after AdapterRemoval, that most of the artefacts are removed. ⚠️ If you turned on
--post_ar_fastq_trimmingyour ‘post-Trimming’ report the statistics after this trimming. There is no separate report for the post-AdapterRemoval trimming.
Sequence Counts
This shows a barplot with the overall number of sequences (x axis) in your raw library after demultiplexing, per file (y-axis). If you have paired end data, you will have one bar for Read 1 (or forward), and a second bar for Read 2 (or reverse). Each entire bar should represent approximately what you requested from the sequencer itself — unless you have your library sequenced over multiple lanes, where it should be what you request divided by the number of lanes it was split over.
A section of the bar will also show an approximate estimation of the fraction of the total number of reads that are duplicates of another. This can derive from over-amplification of the library, or lots of single adapters. This can be later checked with the Deduplication check. A good library and sequencing run should have very low amounts of duplicates reads.
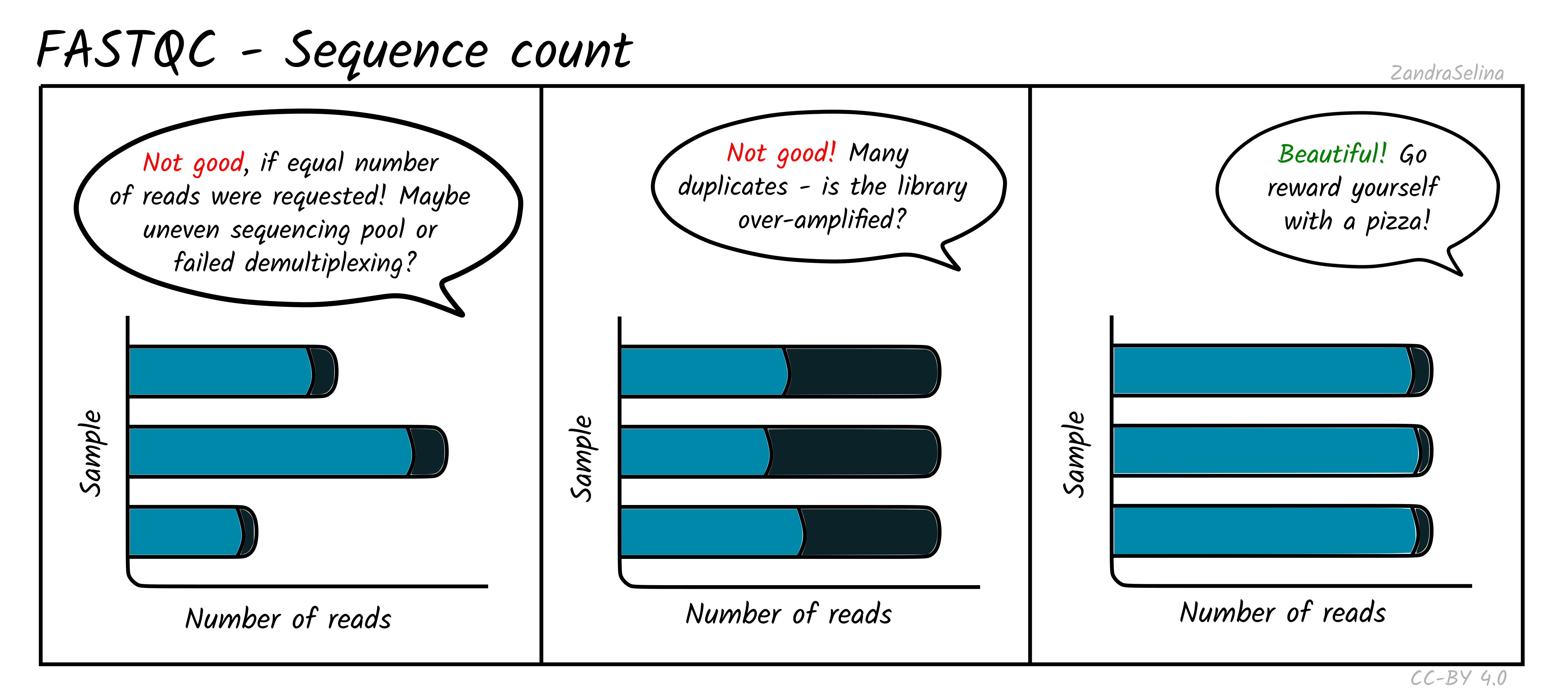
Sequence Quality Histograms
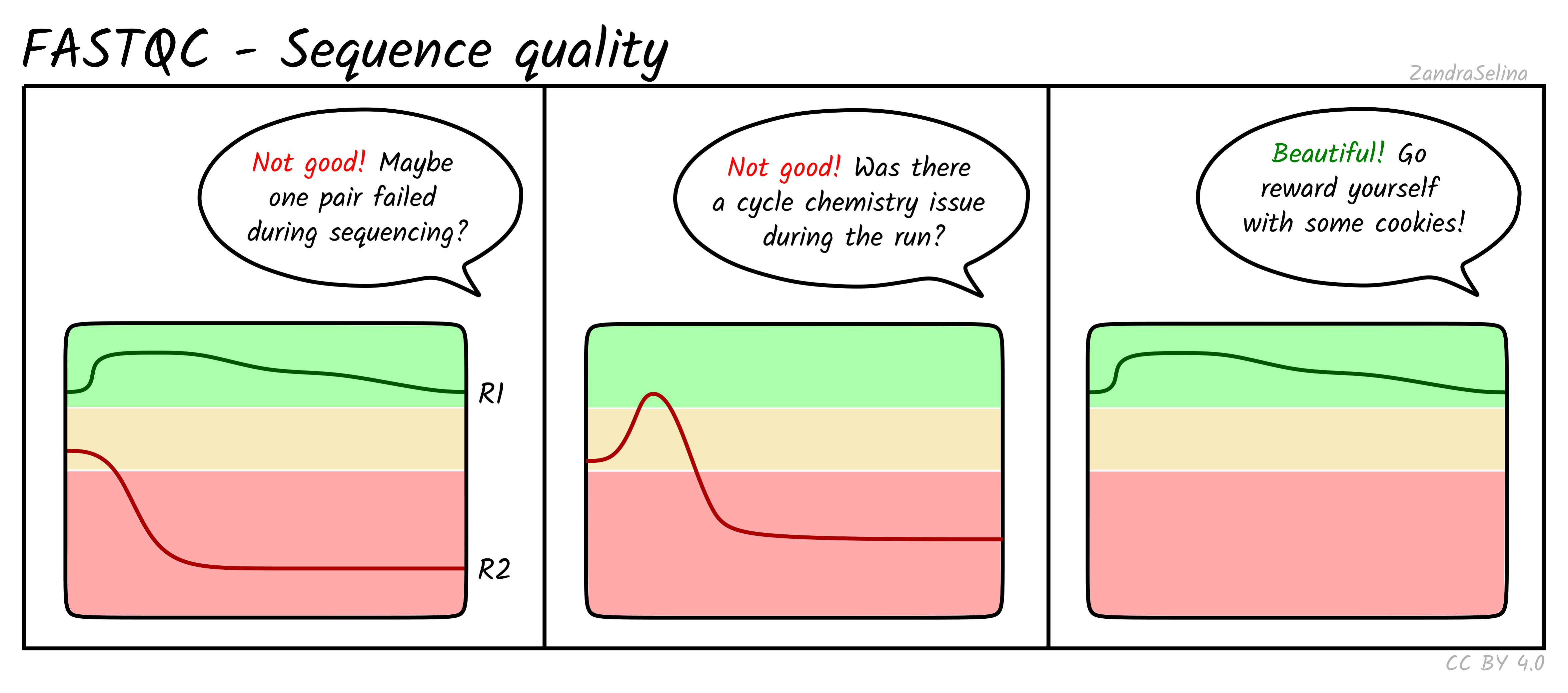
This line plot represents the Phred scores across each base pair of all the reads. The x-axis is the base position across each read, and the y-axis is the average base-calling score (Phred-scaled) of the nucleotides across all reads. Again, this is per FASTQ file (i.e. forward/reverse and/or lanes separately). The background colours represent approximate ranges of quality, with green section being acceptable quality, orange is dubious and red is bad.
You will often see that the first 5 or so bases have slightly lower quality than the rest of the read as this the calibration steps of the machine. The bulk of the read should then stay ~35. Do not worry if you see the last 10-20 bases of reads do often have lower quality base calls that the middle of the read, as the sequencing reagents start to deplete during these cycles (e.g. making nucleotide fluorescence weaker). Furthermore, the reverse reads of sequencing data will often be even lower at ends than forward reads for the same reason.
Things to watch out for:
- all positions having Phred scores less than 27
- a sharp drop-off of quality early in the read
- for paired-end data, if either R1 or R2 is significantly lower quality across the whole read compared to the complementary read.
Per Sequence Quality Scores
This is a further summary of the previous plot. This is a histogram of the overall read quality (compared to per-base, above). The x axis is the mean read-quality score (summarising all the bases of the read in a single value), and the y-axis is the number of reads with this Phred score. You should see a peak with the majority of your reads between 27-35.

Things to watch out for:
- bi-modal peaks which suggests artefacts in some of the sequencing cycles
- all peaks being in orange or red sections which suggests an overall bad sequencing run (possibly due to a faulty flow-cell).
Per Base Sequencing Content
This is a heatmap which shows the average percentage of C, G, T, and A nucleotides across ~4bp bins across all reads.
You expect to see whole heatmap to be a relatively equal block of colour (normally black), representing an equal mix of A, C, T, G colors (see legend).

Things to watch out for:
- If you see a particular colour becoming more prominent this suggests there is an over-representation of those bases at that base-pair range across all reads (e.g. 20-24bp). This could happen if you have lots of PCR duplicates, or poly-G tails from Illumina NextSeq/NovaSeq 2-colour chemistry data (where no fluorescence can mean both G or ‘no-call’).
If you see Poly-G tails, we recommend to turn on FastP poly-G trimming with EAGER. See the ‘running’ documentation page for details.
Per Sequence GC Content
This line graph shows the number percentage reads (y-axis) with an average percent GC content (y-axis). In ‘isolate’ samples (i.e. majority of the reads should be from the host species of the sample), this should be represented by a sharp peak around the average percent GC content of the reference genome. In metagenomic contexts this should be a wide flat distribution with a mean around 50%, however this can be highly different for other types of data.

Things to watch out for:
- If you see particularly high percent GC content peak with NextSeq/NovaSeq data, you may have lots of PCR duplicates, or poly-G tails from Illumina NextSeq/NovaSeq 2-colour chemistry data (where no fluorescence can mean both G or ‘no-call’). Consider re-running nf-core/eager using the poly-G trimming option from
fastpSee the ‘running’ documentation page for details.
Per Base N Content
This line graph shows you the average numbers of Ns found across all reads of a sample. Ns can be caused for a variety of reasons such as low-confidence base call, or the base has been masked. The lines should be very low (as close to 0 as possible) and generally be flat across the whole read. Increases in Ns may reflect in HiSeq data issues of the last cycles running out of chemistry.

NB: Publicly downloaded data may have extremely high N contents across all reads. These normally come from ‘masked’ reads that may have originally be, for example, from a human sample for microbial analysis where the consent for publishing of the host DNA was not given. In these cases you do not need to worry about this plot.
Sequence Duplication Levels
This plot is some-what similar to looking at duplication rate or ‘cluster factor’ of mapped reads. In this case however FastQC takes the sequences of the first 100 thousand reads of a library, and looks to see how often a read sequence is repeated in the rest of the library.
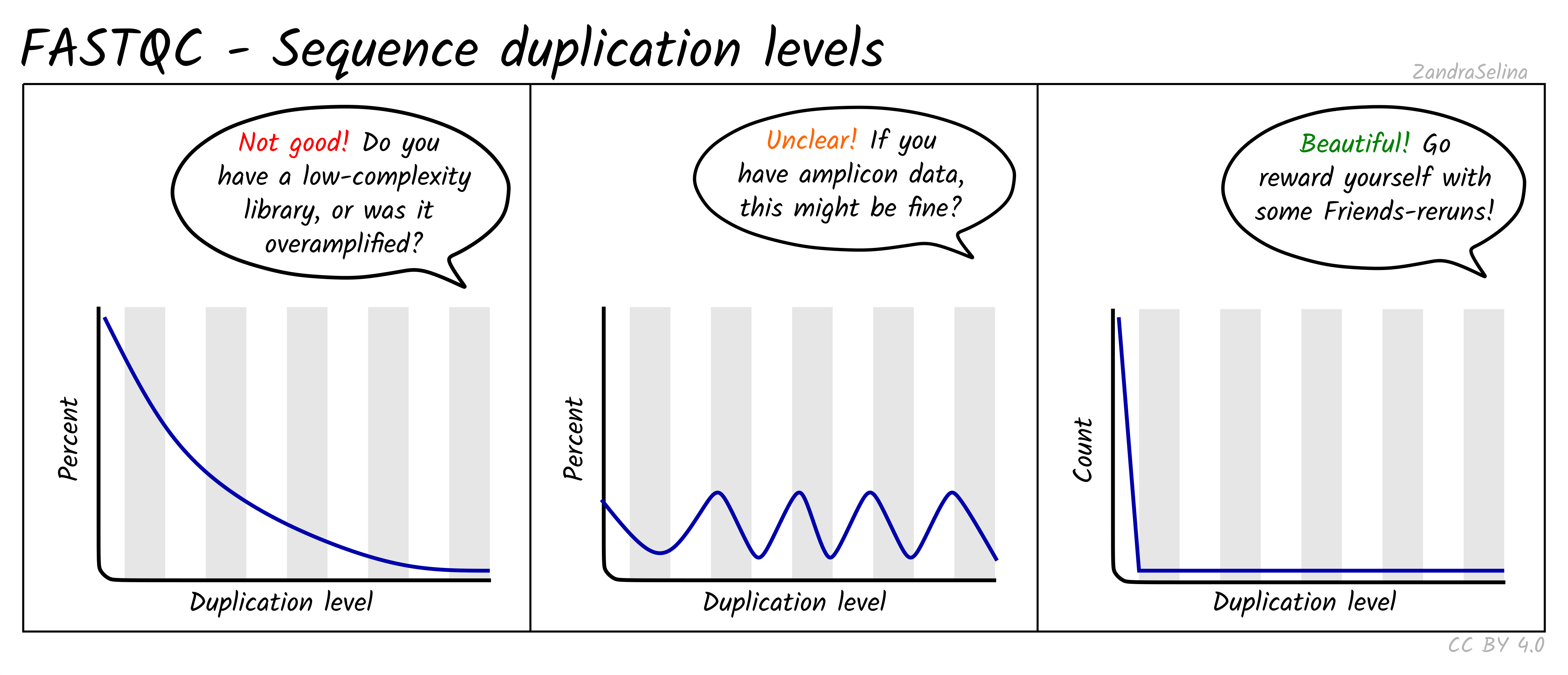
A good library should have very low rates of duplication (vast majority of reads having a duplication rate of 1) — suggesting ‘high complexity’ or lots of unique reads and useful data. This is represented as a steep drop in the line plot and possible a very small curve at about a duplication rate of 2 or 3 and then remaining at ~0 for higher duplication rates.
Note that good libraries may sometimes have small peaks at high duplication levels. This maybe due to free-adapters (with no inserts), or mono-nucleotide reads (e.g. GGGGG in NextSeq/NovaSeq data).
Bad libraries which have extremely low input DNA (so during amplification the same molecules been amplified repeatedly), or a good library that has been erroneously over-amplified will show very high duplication levels — so a very slowly decreasing curve. Alternatively, if your library construction failed and many adapters were not ligated to insert molecules, a high duplication rate may be caused by these free-adapters (see ‘Overrepresented sequences’ for more information).
NB: amplicon libraries such as for 16S rRNA analysis may appear here as having high duplication rates and these peaks can be ignored. This can be verified if no contaminants are found in the ‘Overrepresented sequences’ section.
Overrepresented sequences
After identifying duplicates (see the previous section), a table will be displayed in the ‘Overrepresented sequences’ section of the report. This is an attempt by FastQC to check to see if the duplicates identified match common contaminants such as free adapters or mono-nucleotide reads.
You can then use this table help inform you in identification where the problem occurred in the construction and sequencing of this library. E.g. if you have high duplication rates but no identified contaminants, this suggests over-amplification of reads rather than left over adapters.
Adapter Content
This plot shows the percentage of reads (y-axis), which has an adapter starting at a particular position along a read (x-axis). There can be multiple lines per sample as each line represents a particular adapter.
It is common in aDNA libraries to see very rapid increases in the proportion of reads with an adapter ‘early on’ in the read, as by nature aDNA molecules are fragmented and very short. Palaeolithic samples can have reads as short as 25bp, so sequences can already start having adapters 25bp into a read.
This can already give you an indication on the authenticity of your library - as if you see very low proportions of reads with adapters this suggests long insert molecules that are less likely to derive from a ‘true’ aDNA library. On the flip-side, if you are working with modern DNA - it can give an indication of over-sonication if you have artificially fragmented your reads to lower than your target molecule length.
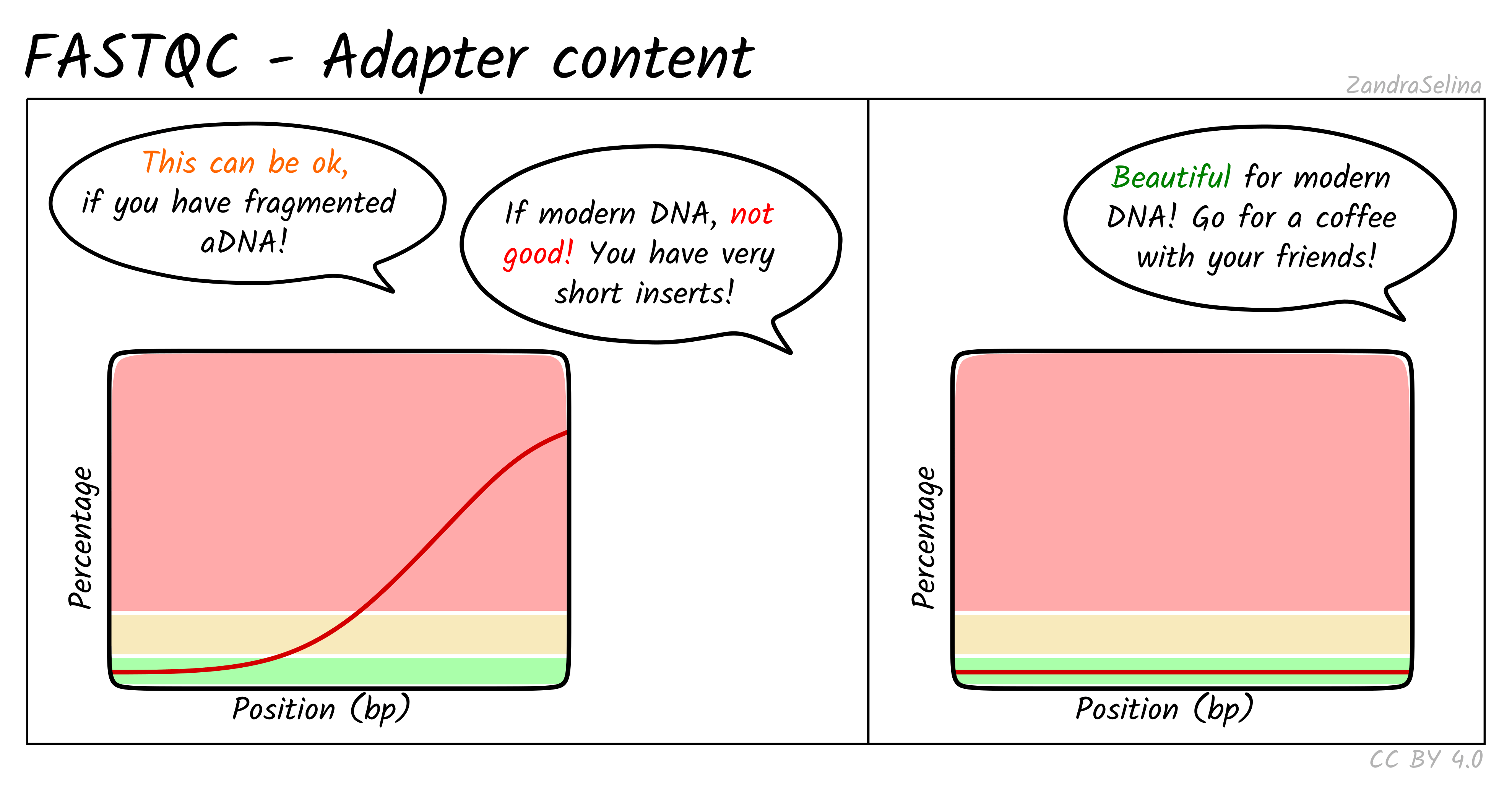
If you have downloaded public data this often is uploaded with adapters already removed, so you can expect a flat distribution straight away.
When comparing pre- and post-AdapterRemoval FASTQC plots of fresh sequencing data (assuming your sequencing center doesn’t already remove adapters), you expect to see something similar to the left panel of the example above pre- adapter removal and the right hand panel post- adapter removal.
FastP
Background
FastP is a general pre-processing toolkit for Illumina sequencing data. In nf-core/eager we currently only use the ‘poly-G’ trimming function. Poly-G tails occur at ends of reads when using two-colour chemistry kits (i.e. in NextSeq and NovaSeq). This occurs as ‘no fluorescence’ is interpreted by the machine; however if the chemistry runs out or the read is shorter than the number of cycles in the kit, you will get at the ends of reads lots of cycles with no nucleotides and these are then recorded as Gs.
While the machine should detect a reduction in base-calling quality, this is not always the case and you will retain these tails in your FASTQ files. This can cause skews in GC content and false positive SNP calls when the reference genome has long mono-nucleotide stretches (typically in larger eukaryotic genomes).
In the case of dual-indexed paired-end sequencing, it is likely poly-G tails are less of an issue as during your AdapterRemoval step, anything passed the adapter will be clipped off anyway. However you can check this under the ‘Per Sequence GC Content’ plot in FastQC.
NB: As you are more likely to see this at the end of the run, in paired-end data you should see all ‘Read 2’ data having a higher GC content distribution than the ‘Read 1’
While the MultiQC report has multiple plots for FastP, we will only look at GC content as that’s the functionality we use currently.
The pipeline will generate the respective output for each supplied FASTQ file.
GC Content
This line plot shows the average GC content (Y axis) across each nucleotide of the reads (X-axis). There are two buttons per read (i.e. 2 for single-end, and 4 for paired-end) representing before and after the poly-G tail trimming.
Before filtering, if you have poly-G tails, you should see the lines going up at the end of the right-hand side of the plot.
After filtering, you should see that the average GC content along the reads is now reduced to around the general trend of the entire read.
Things to look out for:
- If you see a distinct GC content increase at the end of the reads, but are not removed after filtering, check to see where along the read the increase seems to start. If it is less than 10 base pairs from the end, consider reducing the overlap parameter
--complexity_filter_poly_g_min, which tells FastP how far in the read the Gs need to go before removing them.
AdapterRemoval
Background
AdapterRemoval a tool that does the post-sequencing clean up of your sequencing reads. It performs the following functions
- ‘Merges’ (or ‘collapses’) forward and reverse reads of Paired End data
- Removes remaining library indexing adapters
- Trims low quality base tails from ends of reads
- Removes too-short reads
In more detail merging is where the same read from the forward and reverse files of a single library (based on the flowcell coordinates), are compared to find a stretch of sequence that are the same. If this overlap reaches certain quality thresholds, the two reads are ‘collapsed’ into a single read, with the base quality scores are updated accordingly accounting for the increase quality call precision.
Adapter removal involves finding overlaps at the 5’ and 3’ end of reads for the artificial NGS library adapters (which connect the DNA molecule insert, and the index), and stretches that match each other are then removed from the read itself. Note, by default AdapterRemoval does not remove ‘internal barcodes’ (between insert and the adapter), so these statistics are not considered.
Quality trimming (or ‘truncating’) involves looking at ends of reads for low-confidence bases (i.e. where the FASTQ Phred score is below a certain threshold). These are then removed remove the read.
Length filtering involves removing any read that does not reach the number of bases specified by a particular value.
You will receive output for each FASTQ file supplied for single end data, or for each pair of merged FASTQ files for paired end data.
Retained and Discarded Reads Plot
These stacked bars plots are unfortunately a little confusing, when displayed in MultiQC. However are relatively straight-forward once you understand each category. They can be displayed as counts of reads per AdapterRemoval read-category, or as percentages of the same values. Each forward(/reverse) file combination are displayed once.
The most important value is the Retained Read Pairs which gives you the final number of reads output into the file that goes into mapping. Note, however, this section of the stack bar includes the other categories displayed (see below) in the calculation.
Other Categories:
- If paired-end, the Singleton [mate] R1(/R2) categories represent reads which were unable to be collapsed, possibly due to the reads being too long to overlap.
- If paired-end, Full-length collapsed pairs are reads which were collapsed and did not require low-quality bases at end of reads to be removed.
- If paired-end, Truncated collapsed pairs are paired-end that were collapsed but did required the removal of low quality bases at the end of reads.
- Discarded [mate] R1/R2 represent reads which were a part of a pair, but one member of the pair did not reach other quality criteria and was discarded. However the other member of the pair is still retained in the output file as it still reached other quality criteria.

For ancient DNA, assuming a good quality run, you expect to see a the vast majority of your reads overlapping because we have such fragmented molecules. Large numbers of singletons suggest your molecules are too long and may not represent true ancient DNA.
If you see high numbers of discarded or truncated reads, you should check your FastQC results for low sequencing quality of that particular run.
Length Distribution Plot
The length distribution plots show the number of reads at each read-length. You can change the plot to display different categories.
- All represent the overall distribution of reads. In the case of paired-end sequencing You may see a peak at the turn around from forward to reverse cycles.
- Mate 1 and Mate 2 represents the length of the forward and reverse read respectively prior collapsing
- Singleton represent those reads that had a one member of a pair discarded
- Collapsed and Collapsed Truncated represent reads that overlapped and able to merge into a single read, with the latter including base-quality trimming off ends of reads. These plots will start with a vertical rise representing where you are above the minimum-read threshold you set.
- Discarded here represents the number of reads that did not each the read length filter. You will likely see a vertical drop at what your threshold was set to.

With paired-end ancient DNA sequencing runs You expect to see a slight increase in shorter fragments in the reverse (R2) read, as our fragments are so short we often don’t reach the maximum number of cycles of that particular sequencing run.
Bowtie2
Background
This module provides information on mapping when running the Bowtie2 aligner. Bowtie2, like bwa, takes raw FASTQ reads and finds the most likely place on the reference genome it derived from. While this module is somewhat redundant with the Samtools (which reports mapping statistics for bwa) and the endorSp.y endogenous DNA value in the general statistics table, it does provide some details that could be useful in certain contexts.
You will receive output for each library. This means that if you use TSV input and have one library sequenced over multiple lanes and sequencing types, these are merged and you will get mapping statistics of all lanes in one value.
Single/Paired-end alignments
This bar plot shows the number of different categories of reads that Bowtie2 was able to align to the reference genome. You will get slightly different plots for Paired-End (PE) and Single-End (SE) data, but they are basically the same.
Ancient DNA samples typically have low endogenous DNA values, as most of the DNA from the sample is from taphonomic sources (burial environment, modern handling etc), so it is normal to get low numbers of mapping reads.
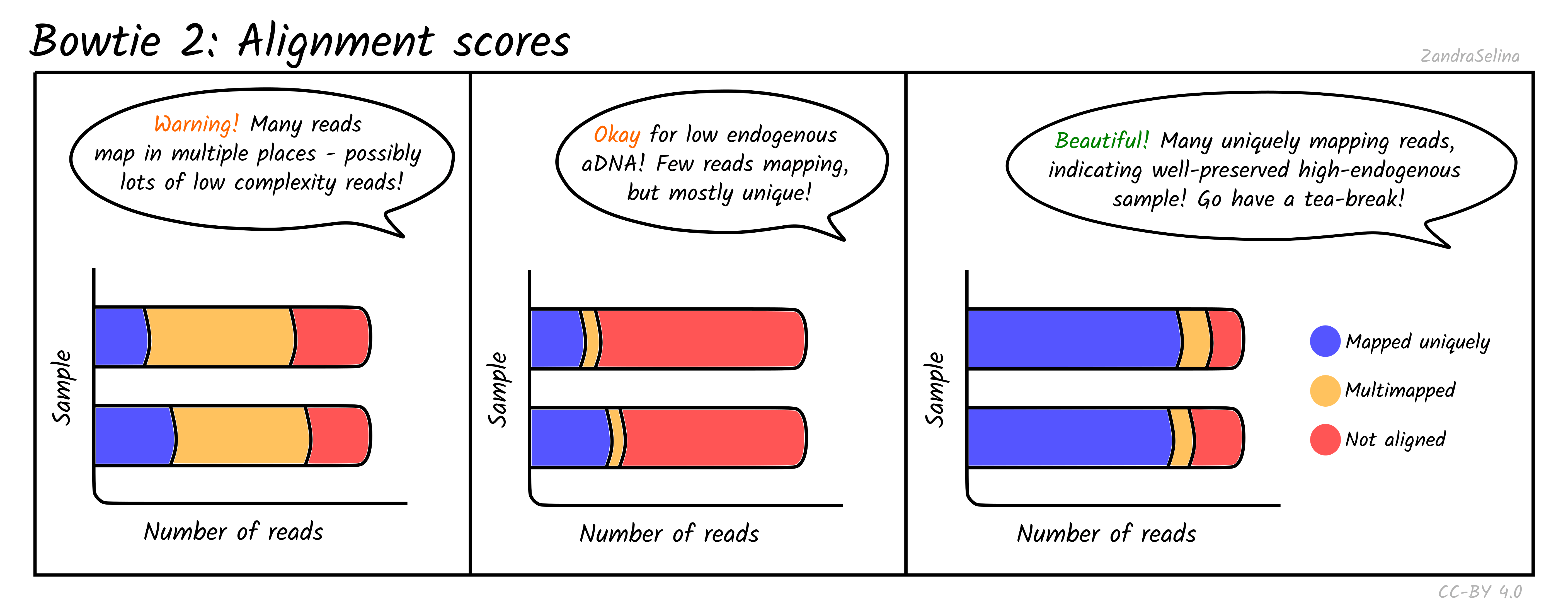
The main additional useful information compared to Samtools is that these plots can inform you how many reads had multiple places on the reference the read could align to. This can occur with low complexity reads or reads derived from e.g. repetitive regions on the genome. If you have large amounts of multi-mapping reads, this can be a warning flag that there is an issue either with the reference genome or library itself (e.g. library construction artefacts). You should investigate cases like this more closely before using the data downstream.
MALT
Background
MALT is a metagenomic aligner (equivalent to BLAST, but much faster). It produces direct alignments of sequencing reads in a reference genome. It is often used for metagenomic profiling or pathogen screening, and specifically in nf-core/eager, of off-target reads from genome mapping.
You will receive output for each library. This means that if you use TSV input and have one library sequenced over multiple lanes and sequencing types, these are merged and you will get mapping statistics of all lanes and sequencing configurations in one value.
Metagenomic Mappability
This bar plot gives an approximation of how many reads in your off-target FASTQ file was able to align to your metagenomic database.
Due to low ‘endogenous’ content of aDNA, and the high biodiversity of modern or environmental microbes that normally exists in archaeological and museum samples, you often will get relatively low mappability percentages.

This can also be influenced by the type of database you supplied — many databases have an over-abundance of taxa of clinical or economic interest, so when you have a large amount of uncharacterised environmental taxa, this may also result in low mappability.
Taxonomic assignment success
In addition to actually being able to align to a given reference sequence, MALT can also allow sequences without a ‘taxonomic’ ID to be included in a database. Furthermore, it utilises a ‘lowest common ancestor’ algorithm (LCA), that can result in a read getting no taxonomic identification (because it can align to multiple reference sequences with equal probability). Because of this, MultiQC also produces a bar plot indicating of the successfully aligned reads (see Metagenomic Mappability above), how many could be assigned a taxon ID.
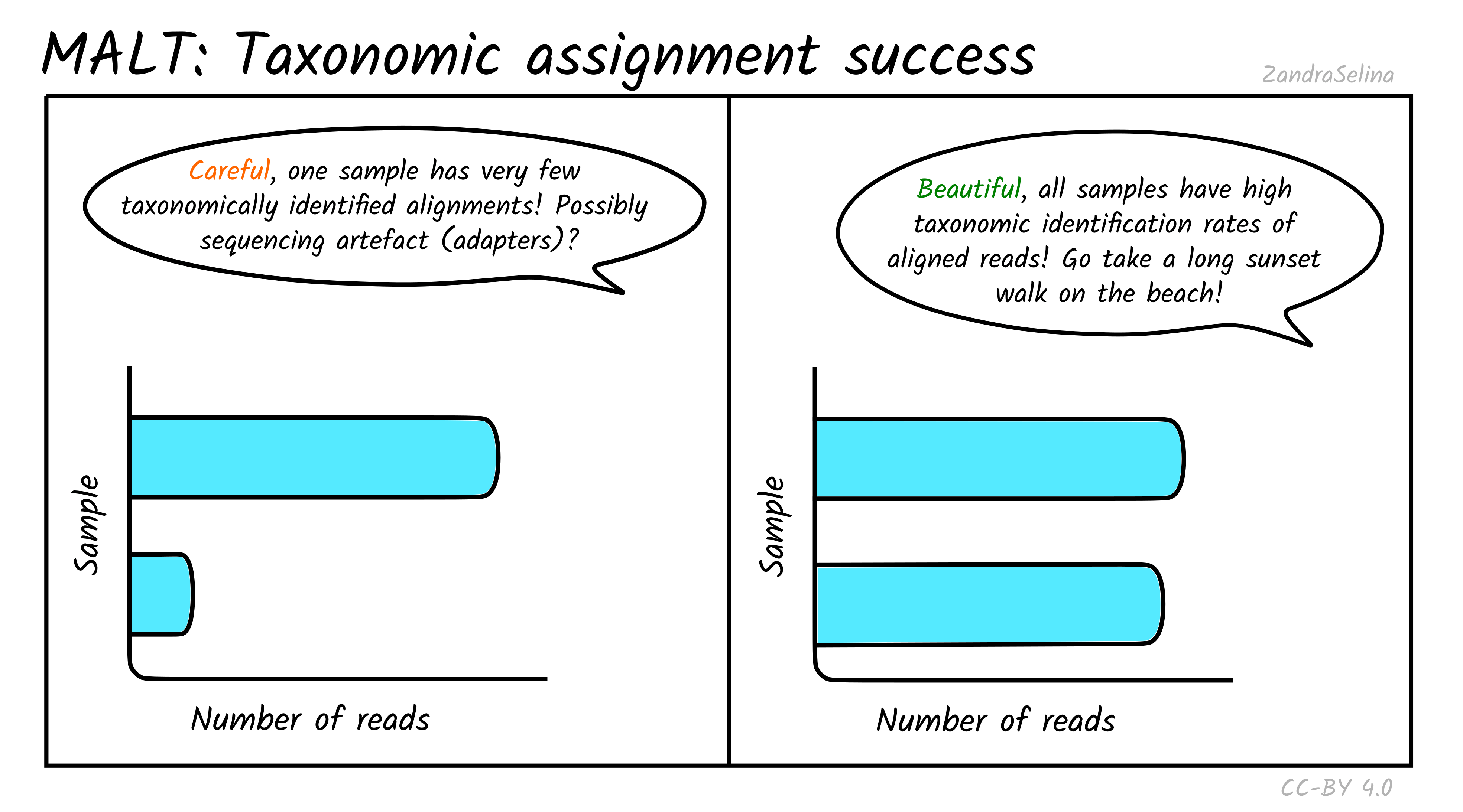
For the same reasons above, you can often get not very many reads being taxonomically assigned when working with aDNA. This can also occur when many of your reads are from conservative regions of genomes and can map onto multiple references. At this point LCA pushes the possible taxon identification so high up the tree, it cannot give a taxonomic assignment.
If you have multiple samples of a similar level of preservation, but one with unusually low numbers of taxonomically assigned reads, it maybe worth investigating what the alignments look like in case there is some sequencing artefact (although it could just be badly preserved and little DNA).
Kraken
Background
Kraken is another metagenomic classifier, but takes a different approach to alignment as with MALT. It uses ‘K-mer similarity’ between reads and references to very efficiently find similar patterns in sequences. It does not however, do alignment — meaning you cannot screen for authentication criteria such as damage patterns and fragment lengths.
It is useful when you do not have large computing power or you want very rapid but rough approximation of the metagenomic profile of your sample.
You will receive output for each library. This means that if you use TSV input and have one library sequenced over multiple lanes and sequencing types, these are merged and you will get mapping statistics of all lanes and sequencing configurations in one value.
Top Taxa
This plot gives you an approximation of the abundance of the five top taxa identified. Typically for ancient DNA, this will be quite a small fraction of taxa, as archaeological and museum samples have a large biodiversity from environmental microbes — therefore a large fraction of ‘unclassified’ can be quite normal.
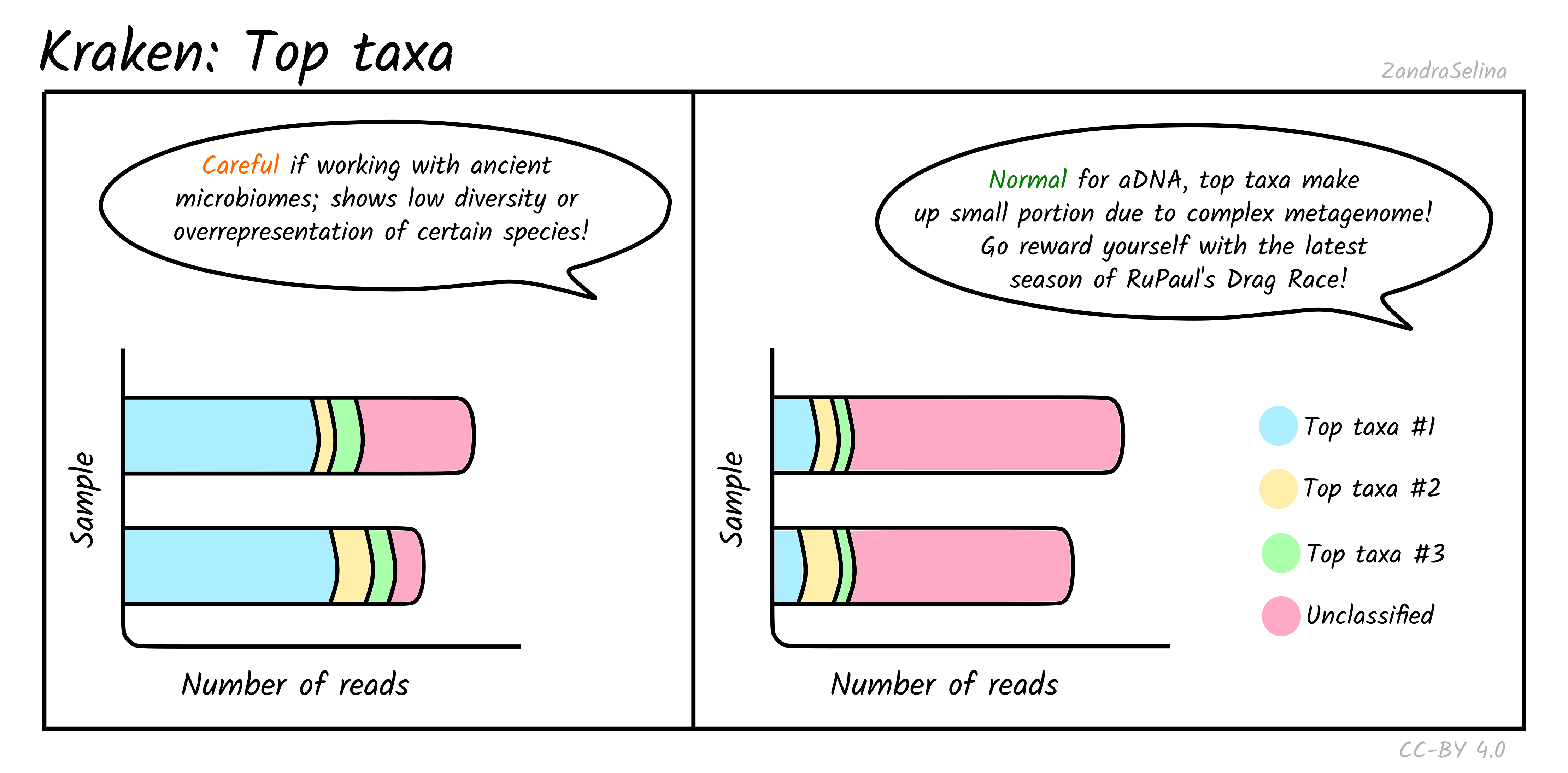
However for screening for specific metagenomic profiles, such as ancient microbiomes, if the top taxa are from your specific microbiome of interest (e.g. looking at calculus for oral microbiomes, or paleofaeces for gut microbiome), this can be a good indicator that you have a well preserved sample. But of course, you must do further downstream (manual!) authentication of these taxa to ensure they are not from modern contamination.
Samtools
Background
This module provides numbers in raw counts of the mapping of your DNA reads to your reference genome.
You will receive output for each library. This means that if you use TSV input and have one library sequenced over multiple lanes and sequencing types, these are merged and you will get mapping statistics of all lanes in one value.
Flagstat Plot
This dot plot shows different statistics, and the number of reads (typically as an multiple e.g. million, or thousands), are represented by dots on the X axis.
In most cases the first two rows, ‘Total Reads’ and ‘Total Passed QC’ will be the same as EAGER (v1) does not do quality control of reads with samtools. This number should normally be the same the number of (clipped, and if paired-end, merged) retained reads coming out of AdapterRemoval.
The third row ‘Mapped’ represents the number of reads that found a place that could be aligned on your reference genome. This is the raw number of mapped reads, prior PCR duplication.
The remaining rows will be 0 when running bwa aln as these characteristics of the data are not considered by the algorithm by default.
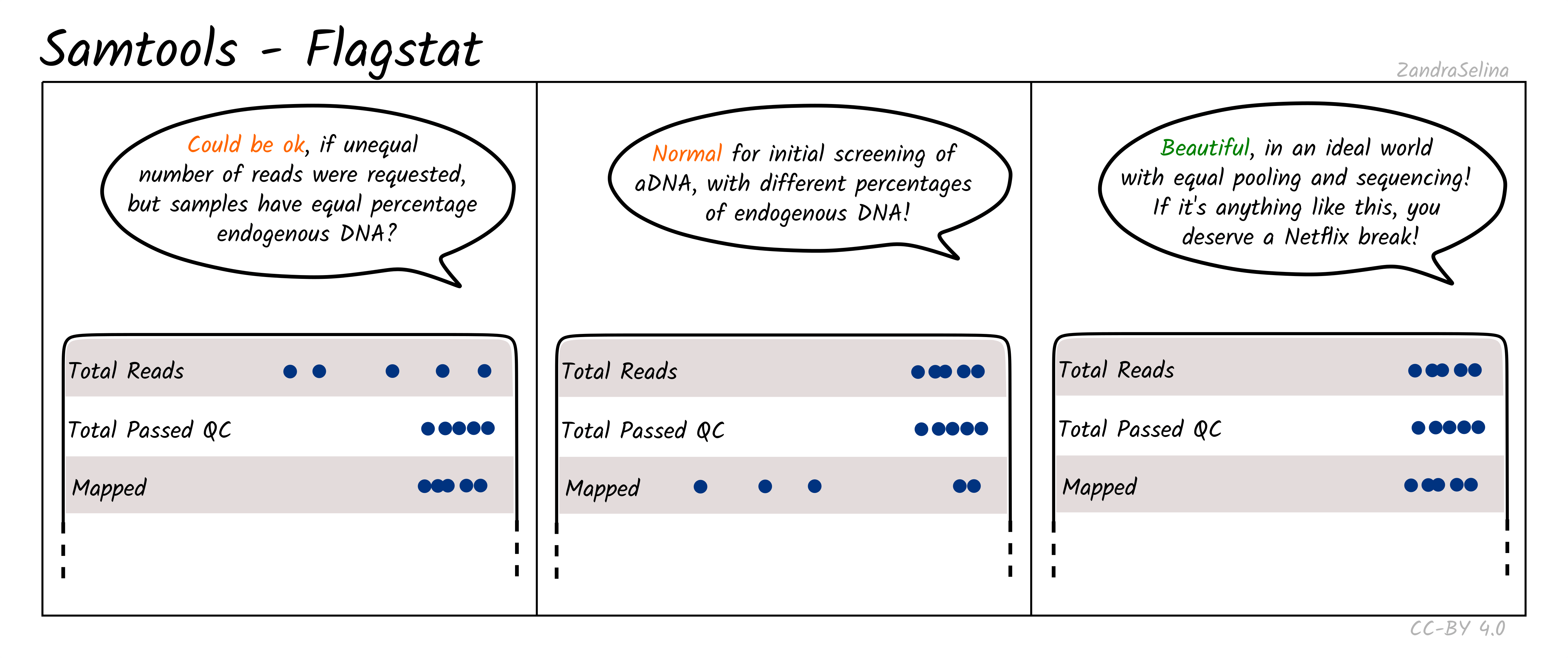
NB: The Samtools (pre-samtools filter) plots displayed in the MultiQC report shows mapped reads without mapping quality filtering. This will contain reads that can map to multiple places on your reference genome with equal or slightly less mapping quality score. To see how your reads look after mapping quality, look at the FastQC reports in the Samtools (pre-samtools filter). You should expect after mapping quality filtering, that you will have less reads.
DeDup
You will receive output for each library. This means that if you use TSV input and have one library sequenced over multiple lanes and sequencing types, these are merged and you will get mapping statistics of all lanes of the library in one value.
Background
DeDup is a duplicate removal tool which searches for PCR duplicates and removes them from your BAM file. We remove these duplicates because otherwise you would be artificially increasing your coverage and subsequently confidence in genotyping, by considering these lab artefacts which are not biologically meaningful. DeDup looks for reads with the same start and end coordinates, and whether they have exactly the same sequence. The main difference of DeDup versus e.g. samtools markduplicates is that DeDup considers both ends of a read, not just the start position, so it is more precise in removing actual duplicates without penalising often already low aDNA data.
DeDup Plot
This stacked bar plot shows as a whole the total number of reads in the BAM file going into DeDup. The different sections of a given bar represents the following:
- Not Removed — the overall number of reads remaining after duplicate removal. These may have had a duplicate (see below).
- Reverse Removed — the number of reads that found to be a duplicate of another and removed that were un-collapsed reverse reads (from the earlier read merging step).
- Forward Removed — the number of reads that found to be a duplicate of another and removed that were an un-collapsed forward reads (from the earlier read merging step).
- Merged Removed — the number of reads that were found to be a duplicate and removed that were a collapsed read (from the earlier read merging step).
Exceptions to the above:
- If you do not have paired end data, you will not have sections for ‘Merged removed’ or ‘Reverse removed’.
- If you use the
--dedup_all_mergedflag, you will not have the ‘Forward removed’ or ‘Reverse removed’ sections.
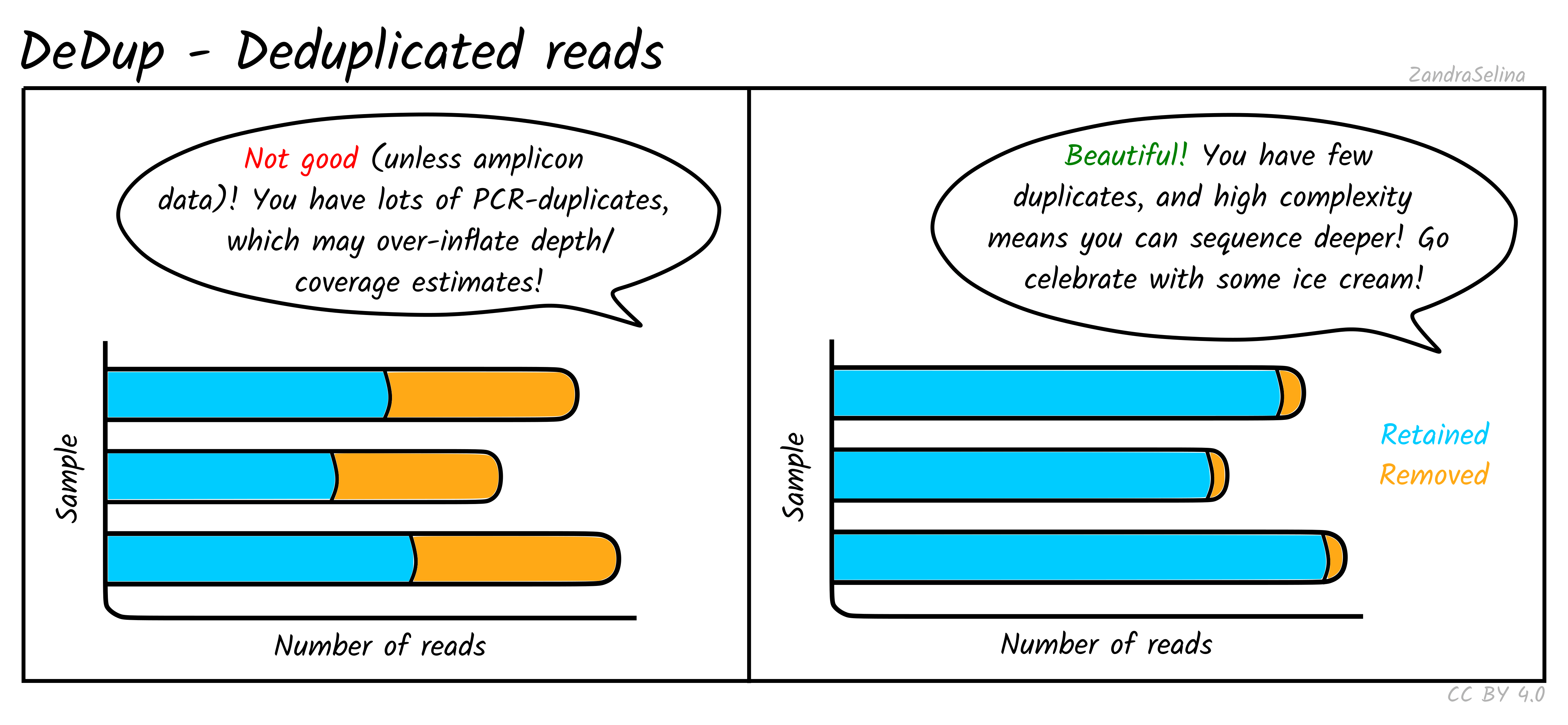
Things to look out for:
- The smaller the number of the duplicates removed the better. If you have a small number of duplicates, and wish to sequence deeper, you can use the preseq module (see below) to make an estimate on how much deeper to sequence.
- If you have a very large number of duplicates that were removed this may suggest you have an over amplified library, or a lot of left-over adapters that were able to map to your genome.
Picard
Background
Picard is a toolkit for general BAM file manipulation with many different functions. nf-core/eager most visibly uses the ‘markduplicates’ tool, for the removal of exact PCR duplicates that can occur during library amplification and results in false inflated coverages (and overly-confident genotyping calls).
Mark Duplicates
The deduplication stats plot shows you how many reads were detected and then removed during deduplication of a mapped BAM file. Well-preserved and constructed libraries will typically have many unique reads and few duplicates. These libraries are often good candidates for deeper sequencing (if required), but low-endogenous DNA libraries that have been over-amplified will have few unique reads and many copies of each read. For better calculations you can see the Preseq module below.
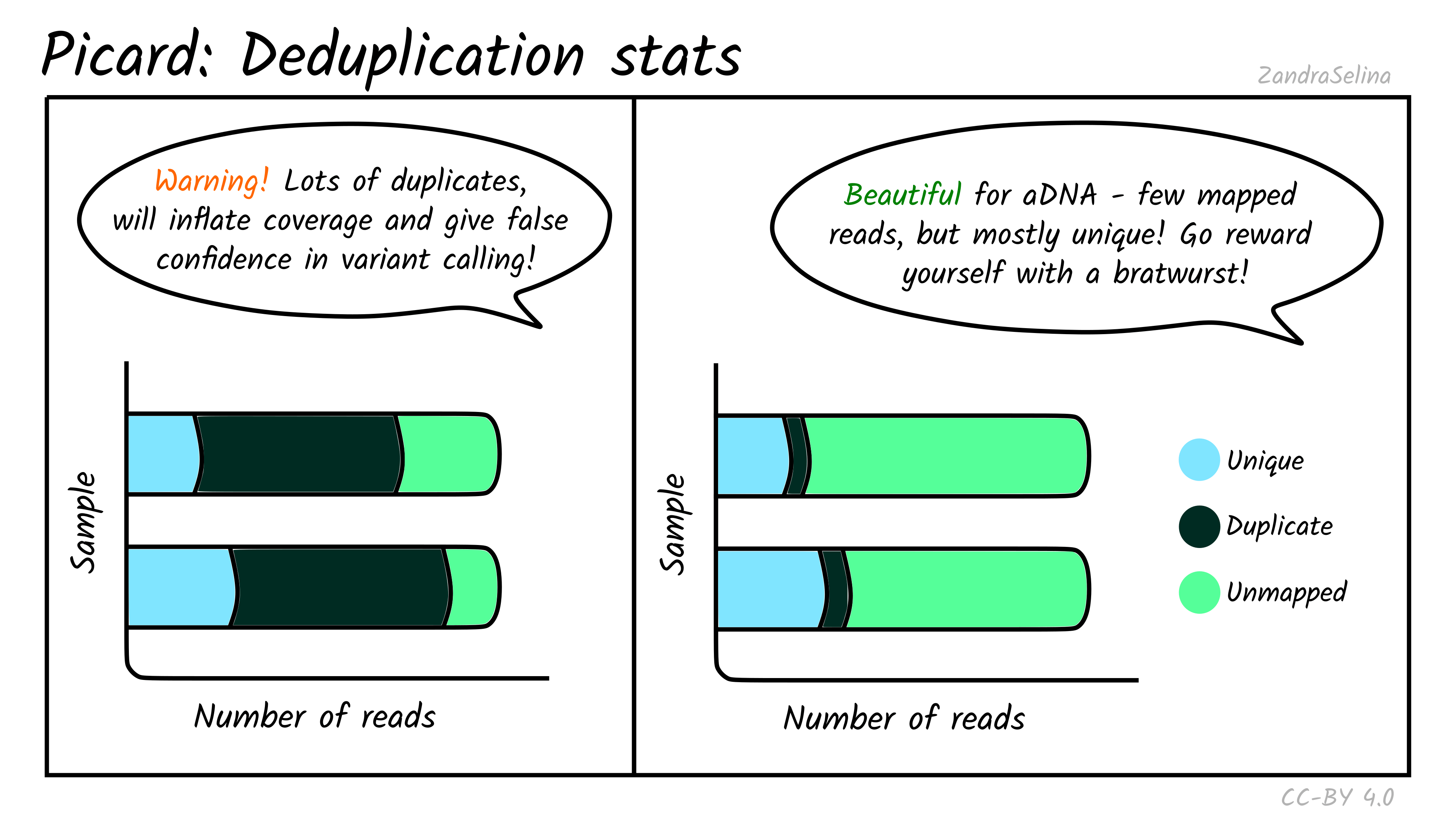
The amount of unmapped reads will depend on whether you have filtered out unmapped reads out not (see the usage/running the pipeline documentation for more information.
Things to look out for:
- The smaller the number of the duplicates removed the better. If you have a smaller number of duplicates, and wish to sequence deeper, you can use the preseq module (see below) to make an estimate on how much deeper to sequence.
- If you have a very large number of duplicates that were removed this may suggest you have an over amplified library, a badly preserved sample with a very low yield, or a lot of left-over adapters that were able to map to your genome.
Preseq
Background
Preseq is a collection of tools that allow assessment of the complexity of the library, where complexity means the number of unique molecules in your library (i.e. not molecules with the exact same length and sequence).
There are two algorithms from the tools we use: c_curve and lc_extrap. The former gives you the expected number of unique reads if you were to repeated sequencing but with fewer reads than your first sequencing run. The latter tries to extrapolate the decay in the number of unique reads you would get with re-sequencing but with more reads than your initial sequencing run.
Due to endogenous DNA being so low when doing initial screening, the maths behind lc_extrap often fails as there is not enough data. Therefore nf-core/eager sticks with c_curve which gives a similar approximation of the library complexity, but is more robust to smaller datasets.
You will receive output for each deduplicated library. This means that if you use TSV input and have one library sequenced over multiple lanes and sequencing types, these are merged and you will get mapping statistics of all lanes of the library in one value.
Complexity Curve
Using the de-duplication information from DeDup, the calculated curve (a solid line) allows you to estimate: at this sequencing depth (on the X axis), how many unique molecules would you have sequenced (along the Y axs). When you start getting DNA sequences that are the mostly same as ones you’ve sequenced before, it is often not cost effective to continue sequencing and is a good point to stop.
The dashed line represents a ‘perfect’ library containing only unique molecules and no duplicates. You are looking for your library stay as close to this line as possible. Plateauing of your curve shows that at that point you would not be getting any more unique molecules and you shouldn’t sequence further than this.
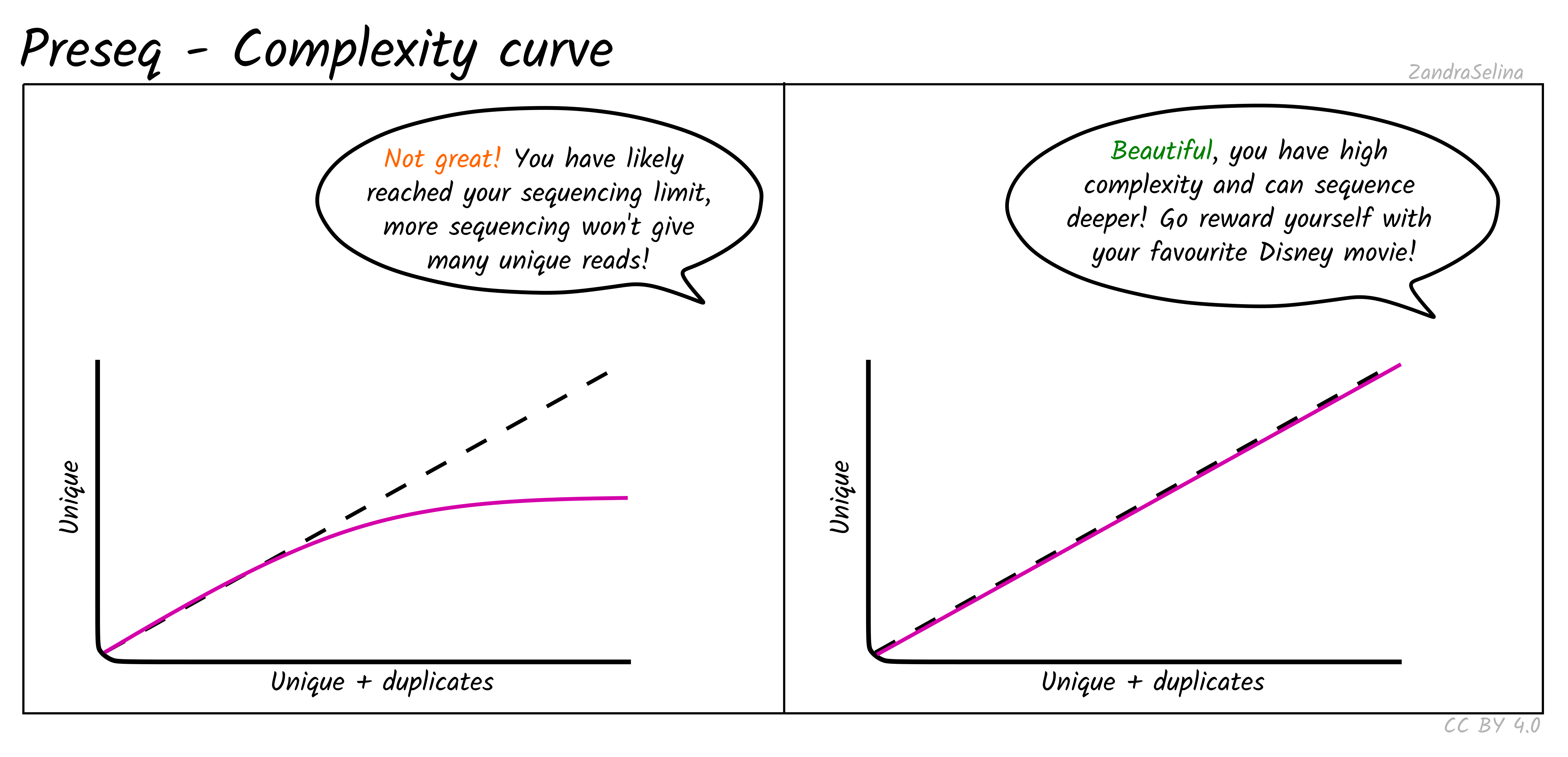
Plateauing can be caused by a number of reasons:
- You have simply sequenced your library to exhaustion
- You have an over-amplified library with many PCR duplicates. You should consider rebuilding the library to maximise data to cost ratio
- You have a low quality library made up of mappable sequencing artefacts that were able to pass filtering (e.g. adapters)
Damage Calculation
Background
DamageProfiler and mapDamage are tools that calculate a variety of standard ‘aDNA’ metrics from a BAM file. The primary plots here are the misincorporation and length distribution plots. Ancient DNA undergoes depurination and hydrolysis, causing fragmentation of molecules into gradually shorter fragments, and cytosine to thymine deamination damage, that occur on the subsequent single-stranded overhangs at the ends of molecules.
Therefore, three main characteristics of ancient DNA are:
- Short DNA fragments
- Elevated G and As (purines) just before strand breaks
- Increased C and Ts at ends of fragments
You will receive output for each deduplicated library. This means that if you use TSV input and have one library sequenced over multiple lanes and sequencing types, these are merged and you will get mapping statistics of all lanes of the library in one value.
Misincorporation Plots
The MultiQC DamageProfiler and mapDamage module misincorporation plots shows the percent frequency (Y axis) of C to T mismatches at 5’ read ends and complementary G to A mismatches at the 3’ ends. The X axis represents base pairs from the end of the molecule from the given prime end, going into the middle of the molecule i.e. 1st base of molecule, 2nd base of molecule etc until the 14th base pair. The mismatches are when compared to the base of the reference genome at that position.
When looking at the misincorporation plots, keep the following in mind:
- As few-base single-stranded overhangs are more likely to occur than long overhangs, we expect to see a gradual decrease in the frequency of the modifications from position 1 to the inside of the reads.
- If your library has been partially-UDG treated, only the first one or two bases will display the misincorporation frequency.
- If your library has been UDG treated you will expect to see extremely-low to no misincorporations at read ends.
- If your library is single-stranded, you will expect to see only C to T misincorporations at both 5’ and 3’ ends of the fragments.
- We generally expect that the older the sample, or the less-ideal preservational environment (hot/wet) the greater the frequency of C to T/G to A.
- The curve will be not smooth then you have few reads informing the frequency calculation. Read counts of less than 500 are likely not reliable.
- If the
mapdamage_downsampleparameter was specified and mapDamage was used for damage calculation, the damage frequency for each base is based only on the specified number of reads.
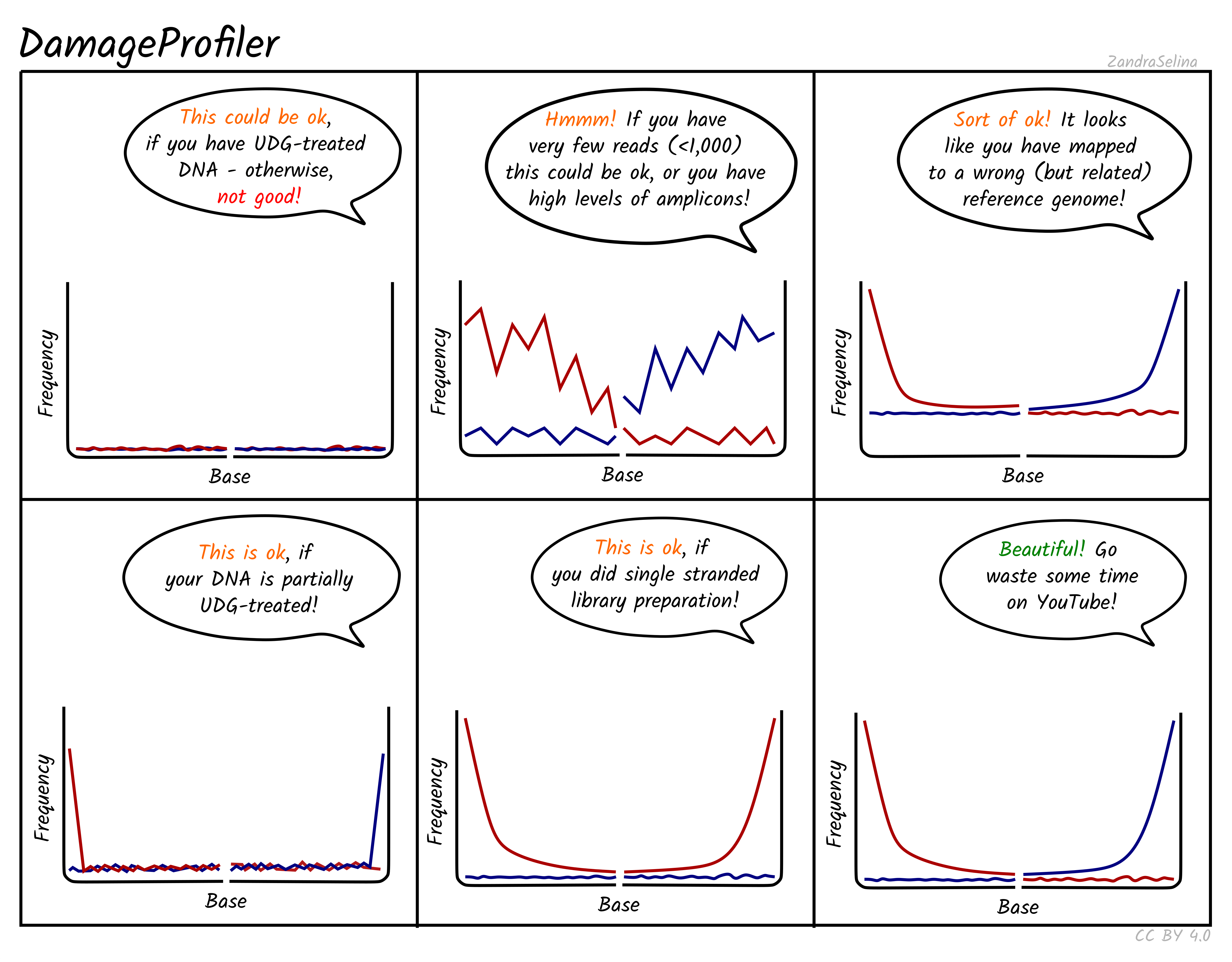
NB: An important difference to note compared to the mapDamage tool, which DamageProfiler is otherwise an exact-re-implementation of, is that the percent frequency on the Y axis is not fixed between 0 and 0.3, and will ‘zoom’ into small values the less damage there is
Length Distribution
The MultiQC DamageProfiler and mapDamage module length distribution plots show the frequency of read lengths across forward and reverse reads respectively.
When looking at the length distribution plots, keep in mind the following:
- Your curves will likely not start at 0, and will start wherever your minimum read-length setting was when removing adapters.
- You should typically see the bulk of the distribution falling between 40-120bp, which is normal for aDNA
- You may see large peaks at paired-end turn-arounds, due to very-long reads that could not overlap for merging being present, however this reads are normally from modern contamination.
- If the
mapdamage_downsampleparameter was specified and mapDamage was used for damage calculation, the length distribution is based only on the specified number of reads.
QualiMap
Background
Qualimap is a tool which provides statistics on the quality of the mapping of your reads to your reference genome. It allows you to assess how well covered your reference genome is by your data, both in ‘fold’ depth (average number of times a given base on the reference is covered by a read) and ‘percentage’ (the percentage of all bases on the reference genome that is covered at a given fold depth). These outputs allow you to make decision if you have enough quality data for downstream applications like genotyping, and how to adjust the parameters for those tools accordingly.
NB: Neither fold coverage nor percent coverage on there own is sufficient to assess whether you have a high quality mapping. Abnormally high fold coverages of a smaller region such as highly conserved genes or un-removed-adapter-containing reference genomes can artificially inflate the mean coverage, yet a high percent coverage is not useful if all bases of the genome are covered at just 1x coverage.
Note that many of the statistics from this module are displayed in the General Stats table (see above), as they represent single values that are not plottable.
You will receive output for each sample. This means you will statistics of deduplicated values of all types of libraries combined in a single value (i.e. non-UDG treated, full-UDG, paired-end, single-end all together).
⚠️ If your library has no reads mapping to the reference, this will result in an empty BAM file. Qualimap will therefore not produce any output even if a BAM exists!
Coverage Histogram
This plot shows on the Y axis the range of fold coverages that the bases of the reference genome are possibly covered by. The Y axis shows the number of bases that were covered at the given fold coverage depth as indicated on the Y axis.
The greater the number of bases covered at as high as possible fold coverage, the better.
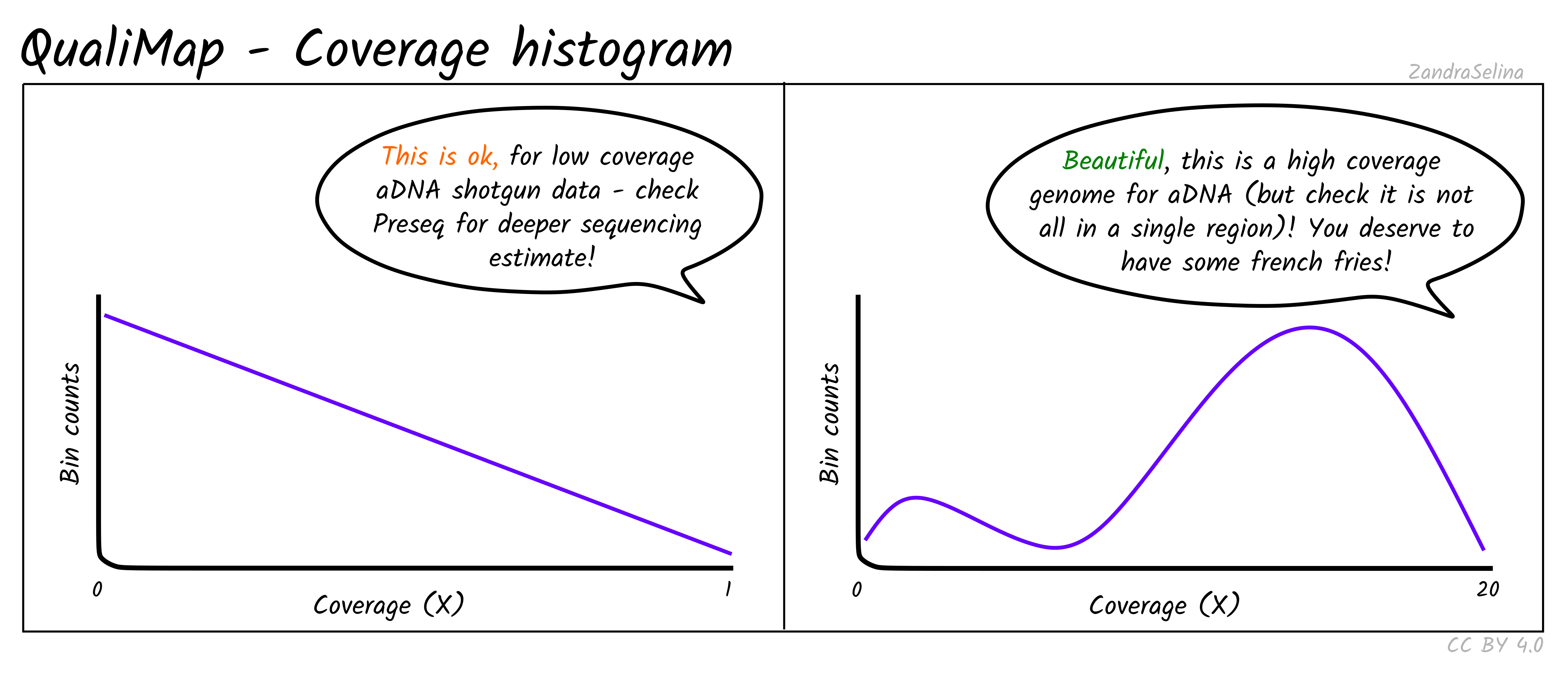
Things to watch out for:
- You will typically see a direct decay from the lowest coverage to higher. A large range of coverages along the X axis is potentially suspicious.
- If you have stacking of reads i.e. a small region with an abnormally large amount of reads despite the rest of the reference being quite shallowly covered, this will artificially increase your coverage. This would be represented by a small peak that is a much further along the X axis away from the main distribution of reads.
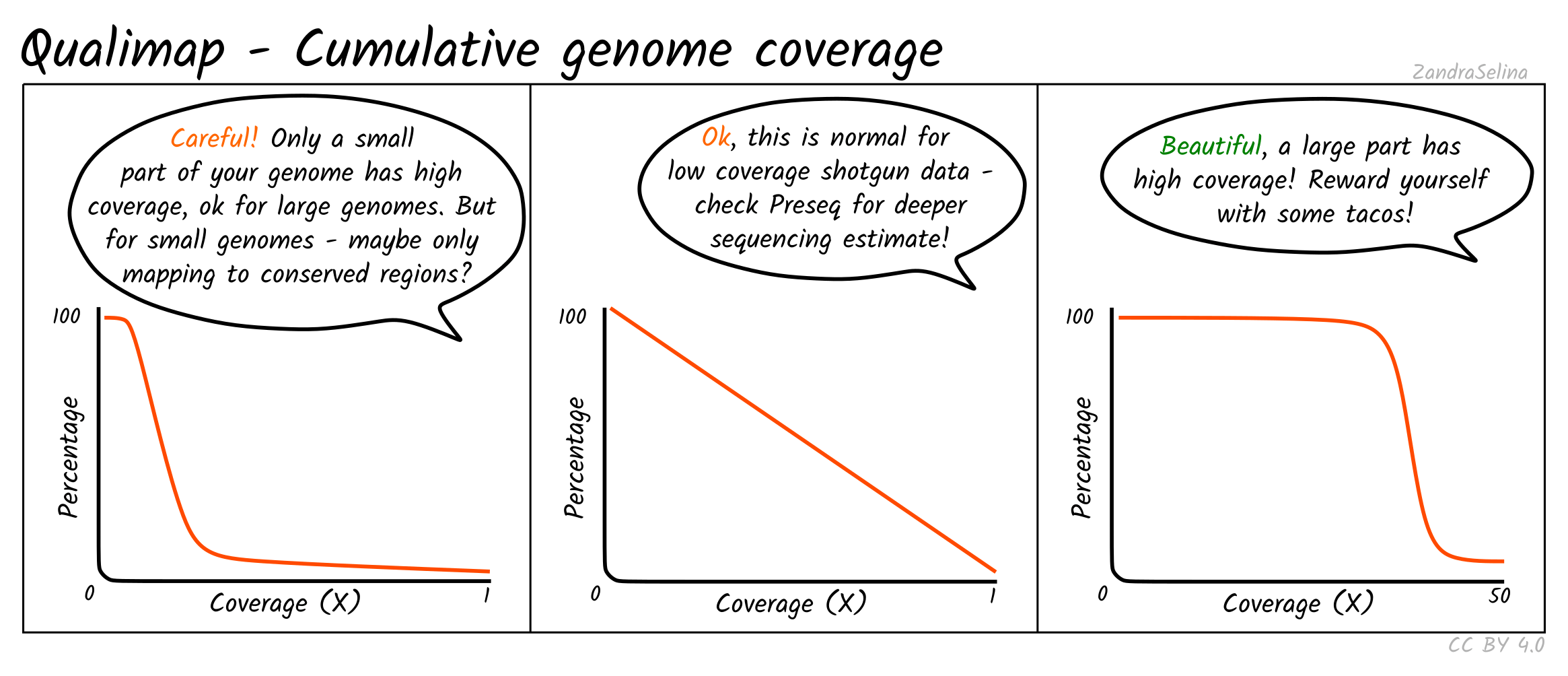
Cumulative Genome Coverage
This plot shows how much of the genome in percentage (X axis) is covered by a given fold depth coverage (Y axis).
An ideal plot for this is to see an increasing curve, representing larger greater fractions of the genome being increasingly covered at higher depth. However, for low-coverage ancient DNA data, you will be more likely to see decreasing curves starting at a large percentage of the genome being covered at 0 fold coverage — something particular true for large genomes such as for humans.
GC Content Distribution
This plot shows the distribution of the frequency of reads at different GC contents. The X axis represents the GC content (i.e the percentage of Gs and Cs nucleotides in a given read), the Y axis represents the frequency.
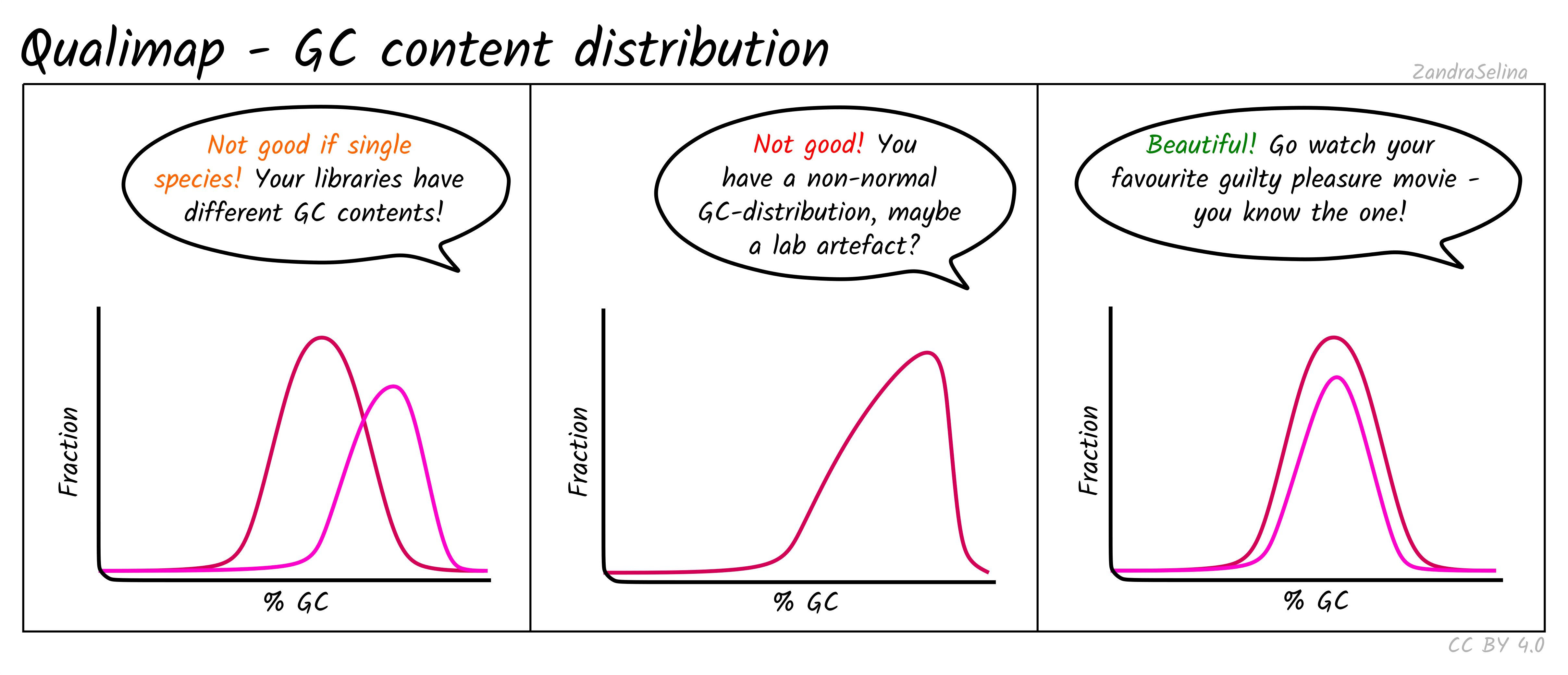
Things to watch out for:
- This plot should normally show a normal distribution around the average GC content of your reference genome.
- Bimodal peaks may represent lab-based artefacts that should be further investigated.
- Skews of the peak to a higher GC content that the reference in Illumina dual-colour chemistry data (e.g. NextSeq or NovaSeq), may suggest long poly-G tails that are mapping to poly-G stretches of your genome. The nf-core/eager trimming option
--complexity_filter_poly_gcan be used to remove these tails by utilising the tool FastP for detection and trimming.
Sex.DetERRmine
Background
Sex.DetERRmine calculates the coverage of your mapped reads on the X and Y chromosomes relative to the coverage on the autosomes (X-/Y-rate). This metric can be thought of as the number of copies of chromosomes X and Y that is found within each cell, relative to the autosomal copies. The number of autosomal copies is assumed to be two, meaning that an X-rate of 1.0 means there are two X chromosomes in each cell, while 0.5 means there is a single copy of the X chromosome per cell. Human females have two copies of the X chromosome and no Y chromosome (XX), while human males have one copy of each of the X and Y chromosomes (XY).
When a bedfile of specific sites is provided, Sex.DetERRmine additionally calculates error bars around each relative coverage estimate. For this estimate to be trustworthy, the sites included in the bedfile should be spaced apart enough that a single sequencing read cannot overlap multiple sites. Hence, when a bedfile has not been provided, this error should be ignored. When a suitable bedfile is provided, each observation of a covered site is independent, and the error around the coverage is equal to the binomial error estimate. This error is then propagated during the calculation of relative coverage for the X and Y chromosomes.
Note that in nf-core/eager this will be run on single- and double-stranded variants of the same library separately. This can also help assess for differential contamination between libraries.
Relative Coverage
Theoretically, males are expected to cluster around (0.5, 0.5) in the produced scatter plot, while females are expected to cluster around (1.0, 0.0). In practice, when analysing ancient DNA, these relative coverage on both axes is slightly lower than expected, and individuals can cluster around (0.45, 0.45) and (0.85, 0.05). As the number of covered sites for an individual gets smaller, the confidence on the estimate becomes lower, because it is increasingly more likely to be affected by randomness in the preservation and sequencing of ancient DNA. Placement of individuals between the male and female clusters can be indicative of low coverage and in some cases contamination, when the contaminant and sampled individuals are of opposite biological sex. Aneuploidy of the sex chromosomes can also be identified with this approach when the placement of an individual in the scatter plot is unexpected. For example, placement of an individual around (1.0, 0.5) despite good genomic coverage is indicative of XXY karyotype (Klinefelter syndrome), while placement around (0.5, 0) could be indicative of karyotype X (Turner’s syndrome).
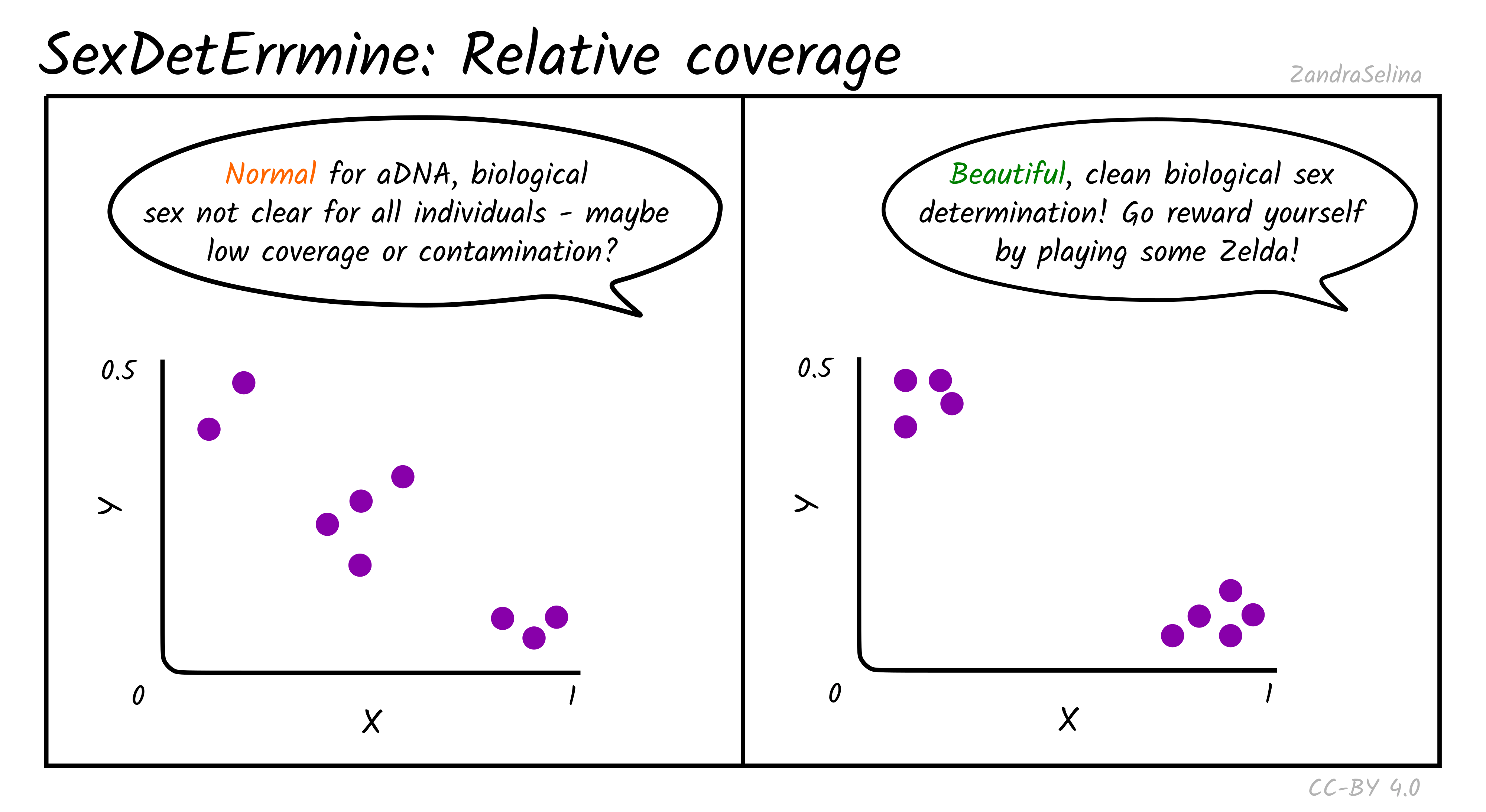
Read Counts
This plot gives you the number of reads mapped onto the autosomes, X or Y chromosomes. When the total number of mapped reads is low, the estimates are more likely to be dominated by random effects, and hence untrustworthy. For well-covered data without any skews, you should see long bars that are comprised mostly of autosomal reads. The edge of the bars in female individuals should be mostly X (some small amounts of Y reads are expected and are usually caused by random mapping on the Y chromosome). In males, the number of X-reads will still be higher (since the X chromosome is longer), but the Y reads should be clearly visible on the rightmost end of the bars. The ratio between the number of sites in each bin should roughly correlate with the difference in length in base pairs of each chromosome type. If this correlation is not observed, your data is skewed towards higher coverage on some chromosomes. This can be expected if you have enriched for a specific set of markers (e.g. Y-chromosome capture), or if the number of reads is too low.
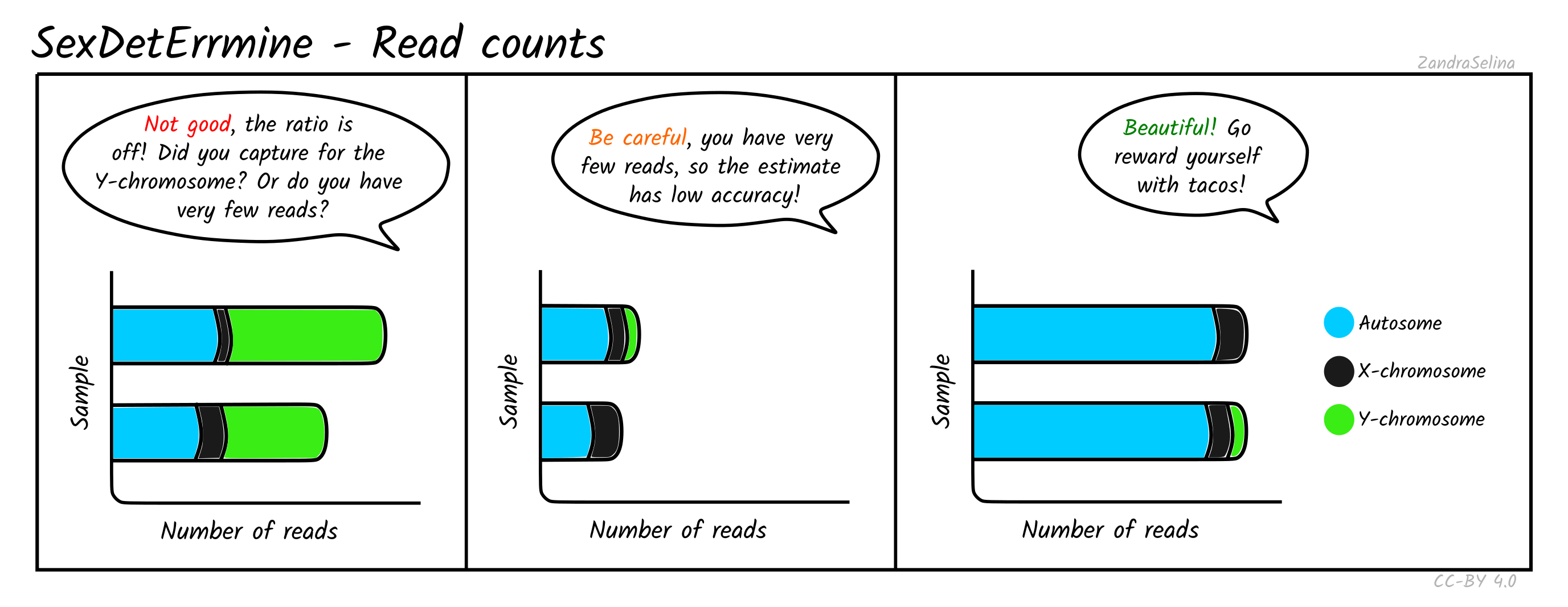
Bcftools
Background
Bcftools is a toolkit for processing and summarising of VCF files, i.e. variant call format files. nf-core/eager currently uses bcftools for the stats functionality. This summarises in a text file a range of statistics about VCF files, produced by GATK and FreeBayes variant callers.
Variant Substitution Types
This stack bar plot shows you the distribution of all types of point-mutation variants away from the reference nucleotide at each position, (e.g. A>C, A>G etc.).
For low-coverage non-UDG treated, non-trimmed nor re-scaled aDNA data, you expect to see a C>T substitutions as the largest category, due to the most common ancient DNA damage being C to T deamination.
Variant Quality
This gives you the distribution of variant-call qualities in your VCF files. Each variant will get given a ‘Phred-scale’ like value that represents the confidence of the variant caller that it has made the right call. The scale is very similar to that of base-call values in FASTQ files (as assessed by FastQC). Distributions that have peaks at higher variant quality scores (>= 30) suggest more confident variant calls. However, in cases of low-coverage aDNA data, these distributions may not be so good.
More detailed explanation of variant quality scores can be seen in the Broad Institute’s GATK documentation.
Indel Distribution
This plot shows you the distribution of the sizes of insertion- and deletions (InDels) in the variant calling (assuming you configured your variant caller parameters to do so). Low-coverage aDNA data often will not have high enough coverage to accurately assess InDels. In cases of high-coverage data of small-genomes such as microbes, large numbers of InDels, however, may indicate your reads are actually from a relative of the reference mapped to - and should be verified downstream.
Variant depths
This plot shows the distribution of depth coverages of each variant called. Typically higher coverage will result in higher quality variant calls (see Variant Quality, above), however in many cases in aDNA these may be low and unequally distributed (due to uneven mapping coverage from contamination).
MultiVCFAnalyzer
Background
MultiVCFanalyzer is a SNP alignment generation tool, that allows further evaluation and filtering of SNP calls made by the GATK UnifiedGenotyper. More specifically it takes one or more VCF files as well as a reference genome, and will allow filtering of SNPs via a variety of metrics and produces a FASTA file with each sample as an entry containing ‘consensus calls’ at each position.
Summary metrics
This table shows the contents of the snpStatistics.tsv file produced by MultiVCFAnalyzer. Descriptions of each column can be seen at the MultiVCFAnalyzer page here.
Call statistics barplot
You can get different variants of the call statistics bar plot, depending on how you configured the MultiVCFAnalyzer options.
If you ran with --min_allele_freq_hom and --min_allele_freq_het set to two different values (left panel A in the figure below), this allows you to assess the number of multi-allelic positions that were called in your genome. Typically MultiVCFAnalyzer is used for analysing smallish haploid genomes (such as mitochondrial or bacterial genomes), therefore a position with multiple possible ‘alleles’ suggests some form of cross-mapping from other taxa or presence of multiple strains. If this is the case, you will need to be careful with downstream analysis of the consensus sequence (e.g. for phylogenetic tree analysis) as you may accidentally pick up SNPs from other taxa/strains — particularly when dealing with low coverage data. Therefore if you have a high level of ‘het’ values (see image), you should carefully check your alignments manually to see how clean your genomes are, or whether you can do some form of strain separation (e.g. by majority/minority calling).

If you ran with --min_allele_freq_hom and --min_allele_freq_het set to the same value, you will have three sections to your bars: SNP Calls (hom), Reference Calls and Discard SNP Call. The overall size of the bars will generally depend on the percentage of the genome covered, meaning less well preserved samples will likely have smaller bars than well-preserved samples. Typically you wish to have a low number of discarded SNP calls, but this can be quite high when you have low coverage data (as many positions may not reach that threshold). The number of SNP calls to reference calls can vary depending on the mutation rate of your target organism.
Output Files
This section gives a brief summary of where to look for what files for downstream analysis. This covers all modules.
Each module has it’s own output directory which sit alongside the MultiQC/ directory from which you opened the report.
reference_genome/: this directory contains the indexing files of your input reference genome (i.e. the variousbwaindices, asamtools’.faifile, and a picard.dict), if you used the--saveReferenceflag.- When masking of the reference is requested prior to running pmdtools, an additional directory
reference_genome/masked_genomewill be found here, containing the masked reference.
- When masking of the reference is requested prior to running pmdtools, an additional directory
fastqc/: this contains the original per-FASTQ FastQC reports that are summarised with MultiQC. These occur in bothhtml(the report) and.zipformat (raw data). Theafter_clippingfolder contains the same but for after AdapterRemoval.adapterremoval/: this contains the log files (ending with.settings) with raw trimming (and merging) statistics after AdapterRemoval. In theoutputsub-directory, are the output trimmed (and merged)fastqfiles. These you can use for downstream applications such as taxonomic binning for metagenomic studies.lanemerging/: this contains adapter-trimmed and merged (i.e. collapsed) FASTQ files that were merged across lanes, where applicable. These files are the reads that go into mapping (when multiple lanes were specified for a library), and can be used for downstream applications such as taxonomic binning for metagenomic studies.post_ar_fastq_trimmed: this containsfastqfiles that have been additionally trimmed after AdapterRemoval (if turned on). These reads are usually that had internal barcodes, or damage that needed to be removed before mapping.mapping/: this contains a sub-directory corresponding to the mapping tool you used, inside of which will be the initial BAM files containing the reads that mapped to your reference genome with no modification (see below). You will also find a corresponding BAM index file (ending in.csior.bai), and if running thebowtie2mapper: a log ending in_bt2.log. You can use these for downstream applications e.g. if you wish to use a different de-duplication tool not included in nf-core/eager (although please feel free to add a new module request on the Github repository’s issue page!).samtools/: this contains two sub-directories.stats/contain the raw mapping statistics files (ending in.stats) from directly after mapping.filter/contains BAM files that have had a mapping quality filter applied (set by the--bam_mapping_quality_thresholdflag) and a corresponding index file. Furthermore, if you selected--bam_discard_unmapped, you will find your separate file with only unmapped reads in the format you selected. Note unmapped read BAM files will not have an index file.deduplication/: this contains a sub-directory calleddedup/, inside here are sample specific directories. Each directory contains a BAM file containing mapped reads but with PCR duplicates removed, a corresponding index file and two stats file..hist.contains raw data for a deduplication histogram used for tools like preseq (see below), and the.logcontains overall summary deduplication statistics.endorSpy/: this contains all JSON files exported from the endorSpy endogenous DNA calculation tool. The JSON files are generated specifically for display in the MultiQC general statistics table and is otherwise very likely not useful for you.preseq/: this contains a.preseqfile for every BAM file that had enough deduplication statistics to generate a complexity curve for estimating the amount unique reads that will be yield if the library is re-sequenced. You can use this file for plotting e.g. inRto find your sequencing target depth.qualimap/: this contains a sub-directory for every sample, which includes a qualimap report and associated raw statistic files. You can open the.htmlfile in your internet browser to see the in-depth report (this will be more detailed than in MultiQC). This includes stuff like percent coverage, depth coverage, GC content and so on of your mapped reads.damageprofiler/: this contains sample specific directories containing raw statistics and damage plots from DamageProfiler. The.pdffiles can be used to visualise C to T miscoding lesions or read length distributions of your mapped reads. All raw statistics used for the PDF plots are contained in the.txtfiles.mapdamage/: this contains sample specific directories containing raw statistics and damage plots from mapDamage. The.pdffiles can be used to visualise C to T miscoding lesions or read length distributions of your mapped reads. All raw statistics used for the PDF plots are contained in the.txtfiles. TheRuntime_log.txtfile contains runtime information.pmdtools/: this contains raw output statistics of pmdtools (estimates of frequencies of substitutions), and BAM files which have been filtered to remove reads that do not have a Post-mortem damage (PMD) score of--pmdtools_threshold.trimmed_bam/: this contains the BAM files with X number of bases trimmed off as defined with the--bamutils_clip_half_udg_left,--bamutils_clip_half_udg_right,--bamutils_clip_none_udg_left, and--bamutils_clip_none_udg_rightflags and corresponding index files. You can use these BAM files for downstream analysis such as re-mapping data with more stringent parameters (if you set trimming to remove the most likely places containing damage in the read).damage_rescaling/: this contains rescaled BAM files from mapDamage. These BAM files have damage probabilistically removed via a bayesian model, and can be used for downstream genotyping.genotyping/: this contains all the (gzipped) genotyping files produced by your genotyping module. The file suffix will have the genotyping tool name. You will have files corresponding to each of your deduplicated BAM files (except pileupcaller), or any turned-on downstream processes that create BAMs (e.g. trimmed bams or pmd tools). If--gatk_ug_keep_realign_bamsupplied, this may also contain BAM files from InDel realignment when using GATK 3 and UnifiedGenotyping for variant calling. When pileupcaller is used to create eigenstrat genotypes, this directory also contains eigenstrat SNP coverage statistics.multivcfanalyzer/: this contains all output from MultiVCFAnalyzer, including SNP calling statistics, various SNP table(s) and FASTA alignment files.sex_determination/: this contains the output for the sex determination run. This is a single.tsvfile that includes a table with the sample name, the number of autosomal SNPs, number of SNPs on the X/Y chromosome, the number of reads mapping to the autosomes, the number of reads mapping to the X/Y chromosome, the relative coverage on the X/Y chromosomes, and the standard error associated with the relative coverages. These measures are provided for each bam file, one row per file. If thesexdeterrmine_bedfileoption has not been provided, the error bars cannot be trusted, and runtime will be considerably longer.nuclear_contamination/: this contains the output of the nuclear contamination processes. The directory contains one*.X.contamination.outfile per individual, as well asnuclear_contamination.txtwhich is a summary table of the results for all individual.nuclear_contamination.txtcontains a header, followed by one line per individual, comprised of the Method of Moments (MOM) and Maximum Likelihood (ML) contamination estimate (with their respective standard errors) for both Method1 and Method2.bedtools/: this contains two files as the output from bedtools coverage. One file contains the ‘breadth’ coverage (*.breadth.gz). This file will have the contents of your annotation file (e.g. BED/GFF), and the following subsequent columns: no. reads on feature, # bases at depth, length of feature, and % of feature. The second file (*.depth.gz), contains the contents of your annotation file (e.g. BED/GFF), and an additional column which is mean depth coverage (i.e. average number of reads covering each position).metagenomic_complexity_filter: this contains the output from filtering of input reads to metagenomic classification of low-sequence complexity reads as performed bybbduk. This will include the filtered FASTQ files (*_lowcomplexityremoved.fq.gz) and also the run-time log (_bbduk.stats) for each sample. Note: there are no sections in the MultiQC report for this module, therefore you must check the._bbduk.statsfiles to get summary statistics of the filtering.metagenomic_classification/: this contains the output for a given metagenomic classifier.- Running MALT will contain RMA6 files that can be loaded into MEGAN6 or MaltExtract for phylogenetic visualisation of read taxonomic assignments and aDNA characteristics respectively. Additional a
malt.logfile is provided which gives additional information such as run-time, memory usage and per-sample statistics of numbers of alignments with taxonomic assignment etc. This will also include gzip SAM files if requested. - Running kraken will contain the Kraken output and report files, as well as a merged Taxon count table. You will also get a Kraken kmer duplication table, in a KrakenUniq fashion. This is very useful to check for breadth of coverage and detect read stacking. A small number of aligned reads (low coverage) and a kmer duplication >1 is usually a sign of read stacking, usually indicative of a false positive hit (e.g. from over-amplified libraries). Kmer duplication is defined as: number of kmers / number of unique kmers. You will find two kraken reports formats available:
- the
*.kreportwhich is the old report format, without distinct minimizer count information, used by some tools such as Pavian - the
*.kraken2_reportwhich is the new kraken report format, with the distinct minimizer count information. - finally, the
*.kraken.outfile are the direct output of Kraken2 - ⚠️ If your sample has no hits, no kraken output files will be created for that sample!
- the
- Running MALT will contain RMA6 files that can be loaded into MEGAN6 or MaltExtract for phylogenetic visualisation of read taxonomic assignments and aDNA characteristics respectively. Additional a
maltextract/: this contains aresultsdirectory in which contains the output from MaltExtract - typically one folder for each filter type, an error and a log file. The characteristics of each node (e.g. damage, read lengths, edit distances - each in different txt formats) can be seen in each sub-folder of the filter folders. Output can be visualised either with the HOPS postprocessing script or MEx-IPAconsensus_sequence/: this contains three FASTA files from VCF2Genome of a consensus sequence based on the reference FASTA with each sample’s unique modifications. The main FASTA is a standard file with bases not passing the specified thresholds as Ns. The two other FASTAS (_refmod.fasta.gz) and (_uncertainity.fasta.gz) are IUPAC uncertainty codes (rather than Ns) and a special number-based uncertainty system used for other downstream tools, respectively.merged_bams/initial: these contain the BAM files that would go into UDG-treatment specific BAM trimming. All libraries of the sample sample, and same UDG-treatment type will be in these BAM files.merged_bams/additional: these contain the final BAM files that would go into genotyping (if genotyping is turned on). This means the files will contain all libraries of a given sample (including trimmed non-UDG or half-UDG treated libraries, if BAM trimming turned on)bcftools: this currently contains a single directory calledstats/that includes general statistics on variant callers producing VCF files as output bybcftools stats. These includethings such as the number of positions, number of transititions/transversions and depth coverage of SNPs etc. These are only produced if--run_bcftools_statsis supplied.