nf-core/nanoseq
Nanopore demultiplexing, QC and alignment pipeline
Introduction
nfcore/nanoseq is a bioinformatics analysis pipeline for Nanopore DNA/RNA sequencing data that can be used to perform basecalling, demultiplexing, QC, alignment, and downstream analysis.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where possible, these processes have been submitted to and installed from nf-core/modules in order to make them available to all nf-core pipelines, and to everyone within the Nextflow community!
On release, automated continuous integration tests run the pipeline on a full-sized dataset obtained from the Singapore Nanopore Expression Consortium on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Pipeline Summary
- Demultiplexing (
qcat; optional) - Raw read cleaning (NanoLyse; optional)
- Raw read QC (
NanoPlot,FastQC) - Alignment (
GraphMap2orminimap2)- Both aligners are capable of performing unspliced and spliced alignment. Sensible defaults will be applied automatically based on a combination of the input data and user-specified parameters
- Each sample can be mapped to its own reference genome if multiplexed in this way
- Convert SAM to co-ordinate sorted BAM and obtain mapping metrics (
samtools)
- Create bigWig (
BEDTools,bedGraphToBigWig) and bigBed (BEDTools,bedToBigBed) coverage tracks for visualisation - DNA specific downstream analysis:
- Short variant calling (
medaka,deepvariant, orpepper_margin_deepvariant) - Structural variant calling (
snifflesorcutesv)
- Short variant calling (
- RNA specific downstream analysis:
- Transcript reconstruction and quantification (
bambuorStringTie2)- bambu performs both transcript reconstruction and quantification
- When StringTie2 is chosen, each sample can be processed individually and combined. After which,
featureCountswill be used for both gene and transcript quantification.
- Differential expression analysis (
DESeq2and/orDEXSeq) - RNA modification detection (
xporeand/orm6anet) - RNA fusion detection (
JAFFAL)
- Transcript reconstruction and quantification (
- Present QC for raw read and alignment results (
MultiQC)
Functionality Overview
A graphical overview of suggested routes through the pipeline depending on the desired output can be seen below.
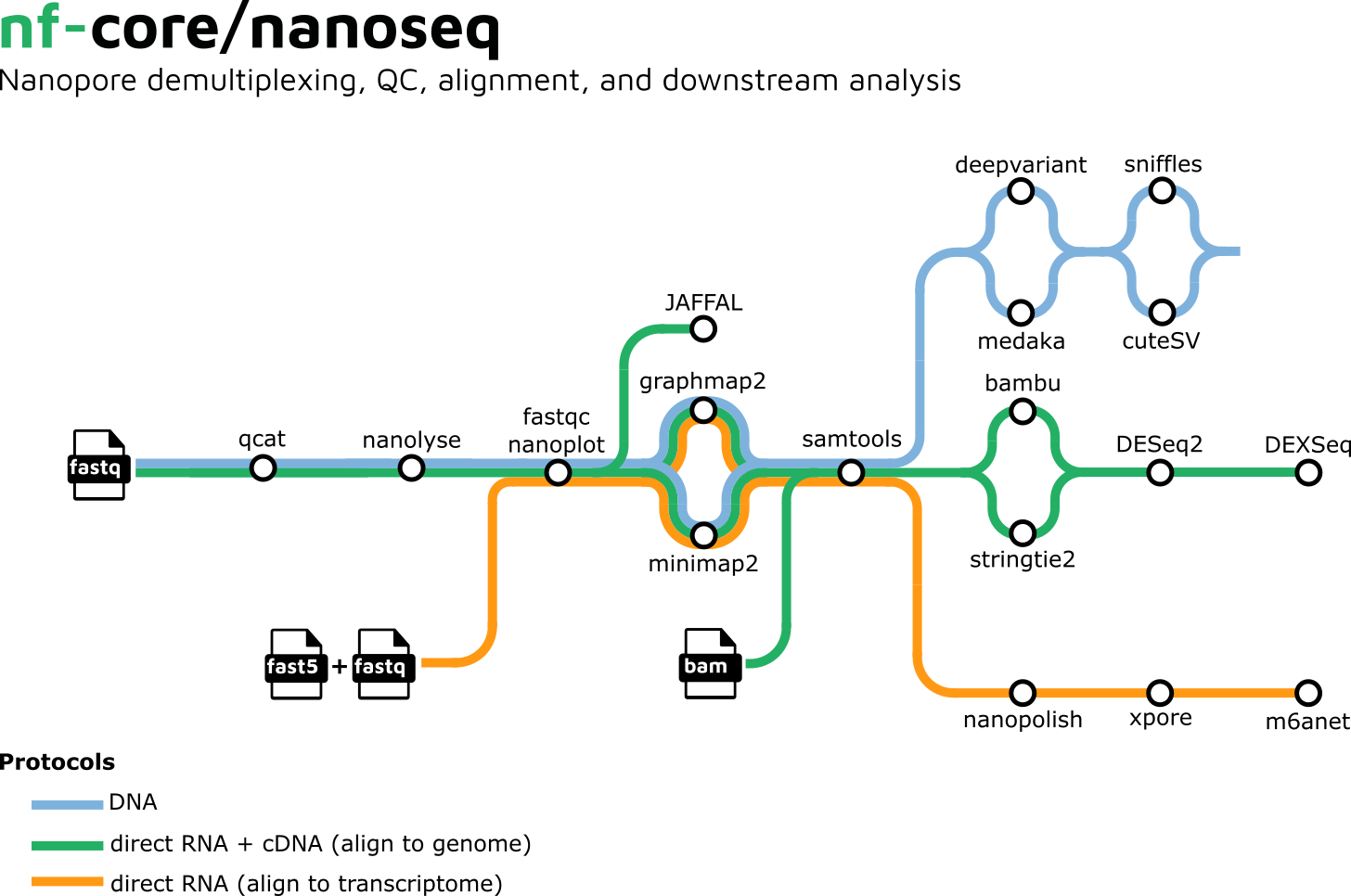
Quick Start
-
Install
Nextflow(>=22.10.1) -
Install any of
Docker,Singularity(you can follow this tutorial),Podman,ShifterorCharliecloudfor full pipeline reproducibility (you can useCondaboth to install Nextflow itself and also to manage software within pipelines. Please only use it within pipelines as a last resort; see docs). -
Download the pipeline and test it on a minimal dataset with a single command:
nextflow run nf-core/nanoseq -profile test,YOURPROFILENote that some form of configuration will be needed so that Nextflow knows how to fetch the required software. This is usually done in the form of a config profile (
YOURPROFILEin the example command above). You can chain multiple config profiles in a comma-separated string.- The pipeline comes with config profiles called
docker,singularity,podman,shifter,charliecloudandcondawhich instruct the pipeline to use the named tool for software management. For example,-profile test,docker. - Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment. - If you are using
singularityand are persistently observing issues downloading Singularity images directly due to timeout or network issues, then you can use the--singularity_pull_docker_containerparameter to pull and convert the Docker image instead. Alternatively, you can use thenf-core downloadcommand to download images first, before running the pipeline. Setting theNXF_SINGULARITY_CACHEDIRorsingularity.cacheDirNextflow options enables you to store and re-use the images from a central location for future pipeline runs. - If you are using
conda, it is highly recommended to use theNXF_CONDA_CACHEDIRorconda.cacheDirsettings to store the environments in a central location for future pipeline runs.
- The pipeline comes with config profiles called
-
Start running your own analysis!
Documentation
The nf-core/nanoseq pipeline comes with documentation about the pipeline usage, parameters and output.
nextflow run nf-core/nanoseq \
--input samplesheet.csv \
--protocol DNA \
--barcode_kit SQK-PBK004 \
-profile <docker/singularity/podman/institute>See usage docs for all of the available options when running the pipeline.
An example input samplesheet for performing both basecalling and demultiplexing can be found here.
Credits
nf-core/nanoseq was originally written by Chelsea Sawyer and Harshil Patel from The Bioinformatics & Biostatistics Group for use at The Francis Crick Institute, London. Other primary contributors include Laura Wratten, Ying Chen, Yuk Kei Wan and Jonathan Goeke from the Genome Institute of Singapore, Christopher Hakkaart from Institute of Medical Genetics and Applied Genomics, Germany, and Johannes Alneberg and Franziska Bonath from SciLifeLab, Sweden.
Many thanks to others who have helped out along the way too, including (but not limited to): @crickbabs, @AnnaSyme, @ekushele.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on Slack (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.