nf-core/rarevariantburden
Pipeline for performing consistent summary count based rare variant burden test, which is useful when we only have sequenced cases data. For example, we can compare the cases against public summary count data, such as gnomAD.
Introduction
nf-core/rarevariantburden (CoCoRV-nf) is a bioinformatics pipeline that performs consistent summary count based rare variant burden test, which is useful when we only have sequenced cases/patients data, no matched control data, here we provided pre-processed and annotated public summary count data, such as gnomAD data, which can be used for rare variant burden test and can be used to identify disease-predisposition genes present in the case study.
Some key features of our pipeline:
- Consistent filtering is applied to make sure the same set of high quality variants are used.
- It can stratify cases into different ethnicity groups, and perform stratified analysis with group-matched control summary counts.
- For recessive models, it can exclude double heterozygous due to high linkage disequilibrium in populations.
- Also provides accurate inflation factor estimate, QQ plot, and powerful FDR control for discrete count data, whose p-value distribution under the null is far from the uniform distribution when the alleles are very rare.
- Also performs sex-stratified analysis if user provides the gender of the input samples
- It supports gnomAD v2 exome (GRCh37) data, and gnomAD v4.1 exome (GRCh38) data, and gnomAD v4.1 genome (GRCh38) data as control.
Pipeline summary
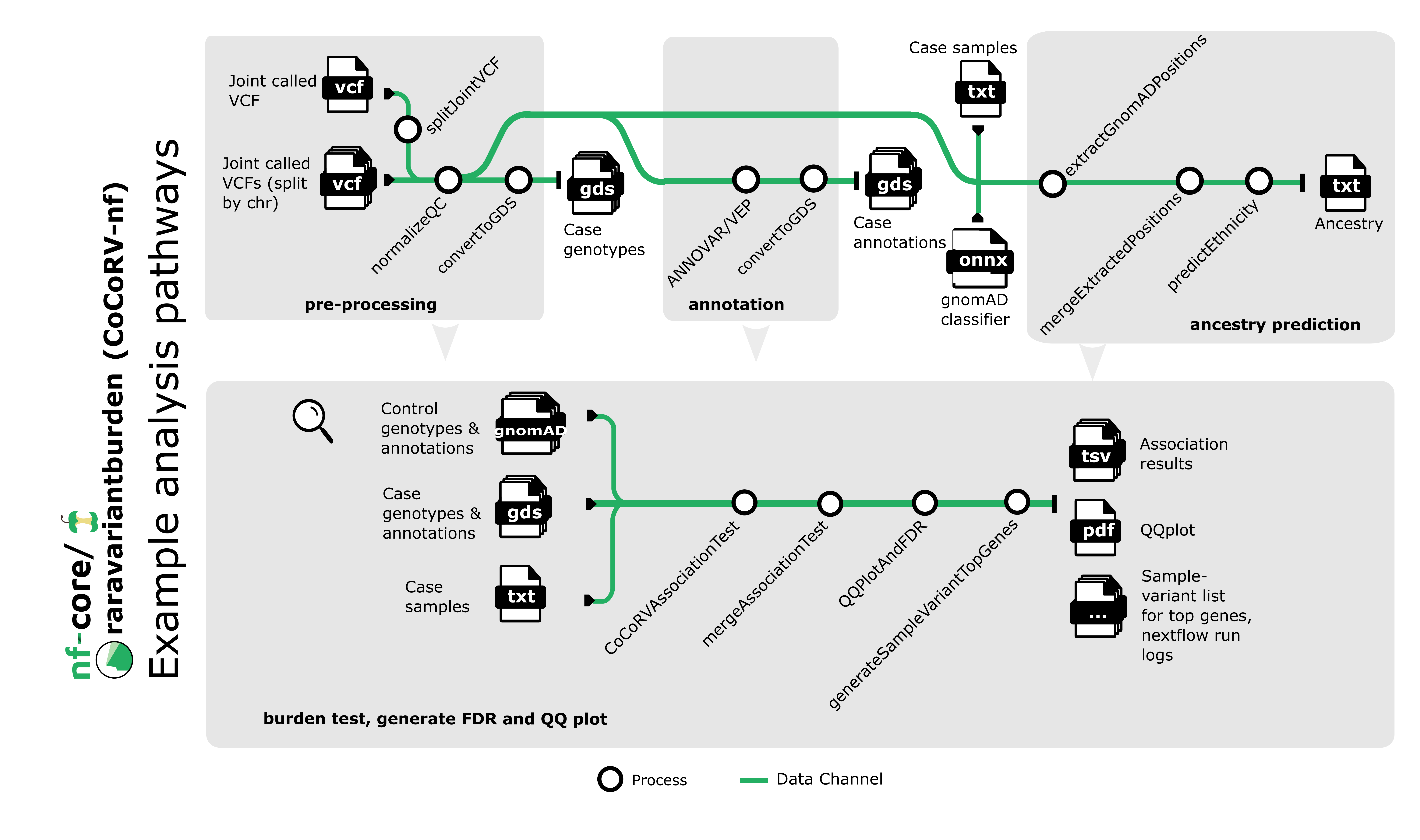
- Split the case joint called and VQSR applied VCF files chromosomewise (Using BCFtools)
- Normalize and QC the splitted case VCF files (Using BCFtools)
- Annotate normalized and QCed VCF files with Annovar and VEP (default annotation tool is ANNOVAR)
- Convert the normalized and annotated VCF files to GDS format, which is easier to process in R (Using R seqarray)
- Predict the ethnicity of the case samples (Using gnomAD random forest classifier)
- Perform association test for each VCF file using our CoCoRV (Consistent summary Count based Rare Variant burden test) R package
- Merge association test results
- Calculate false positive rate (FDR) from merged results, plot QQ plot and lambda value using different R libraries
- For top K genes, generate the list of samples and associated variants along with the annotations for the variants, this list will help the users to further check the top genes and their variants
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare the joint called and VQSR applied VCF file from your case study. You can use nf-core/sarek pipeline’s GATK joint calling module to prepare a joint called and VQSR applied VCF file from your sample VCF files. You also need to prepare a text file containing sample IDs, one sample ID per line.
For control data, you need to download the control data from our Amazon AWS s3 bucket. We provide 3 different control datasets, For build GRCh37, we have gnomADv2exome data, for build GRCh38, we have gnomADv4.1exome and gnomADv4.1genome data as controls.
As the control data is a huge dataset, it is better to use Amazon AWS command line tool aws-cli to download the data.
After installing aws-cli, you can use “aws s3” command to list any s3 bucket folder, or download any folder or files from s3. You will find the s3 commands list in here.
Here are the s3 bucket paths of the 3 gnomAD control datasets:
- s3://cocorv-resource-files/gnomADv2exome/
- s3://cocorv-resource-files/gnomADv4.1exome/
- s3://cocorv-resource-files/gnomADv4.1genome/
To download the data, you need to run following command:
cd /local-dir-path-where-you-want-download/
aws s3 cp s3://cocorv-resource-files/gnomADv2exome/ . --recursiveYou can check all resource files for our pipeline using this command:
aws s3 ls s3://cocorv-resource-files/You also need to download the annovar and VEP resource folders for running Annovar and VEP annotation.
Here are the s3 bucket paths of the annotation tool datasets:
- s3://cocorv-resource-files/annovarFolder/
- s3://cocorv-resource-files/vepFolder/
Now, you can run the pipeline by downloading the code base from github and using the following command:
nextflow run rarevariantburden/main.nf \
-profile <docker/singularity/...>,<institute> \
--caseJointVCF <jointVCF.vcf.gz> \
--caseSample <sampleList.txt> \
--controlDataFolder <controldataFolder> \
--annovarFoler <annovarFolder> \
--vepFolder <vepFolder> \
--reference <GRCh37/GRCh38> \
--gnomADVersion <v2exome/v4exome/v4genome> \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
AWSHealthOmics Implementation
For implementing our pipeline on AWS HealthOmics cloud platform, please refere to our AWS documentation.
DNAnexus Implementation
For implementing our pipeline on DNAnexus cloud platform, please refere to our DNAnexus documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/rarevariantburden (CoCoRV-nf) is written by Saima Sultana Tithi (saimasultana.tithi@stjude.org), St. Jude Children’s Research Hospital, Memphis, TN, USA and Wenan Chen (chen.wenan@mayo.edu), Mayo Clinic, Rochester, MN, USA.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #rarevariantburden channel (you can join with this invite).
Citations
To learn more about the original CoCoRV tool, please look at our paper published in Nature Communications Pubmed link.
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.