Introduction
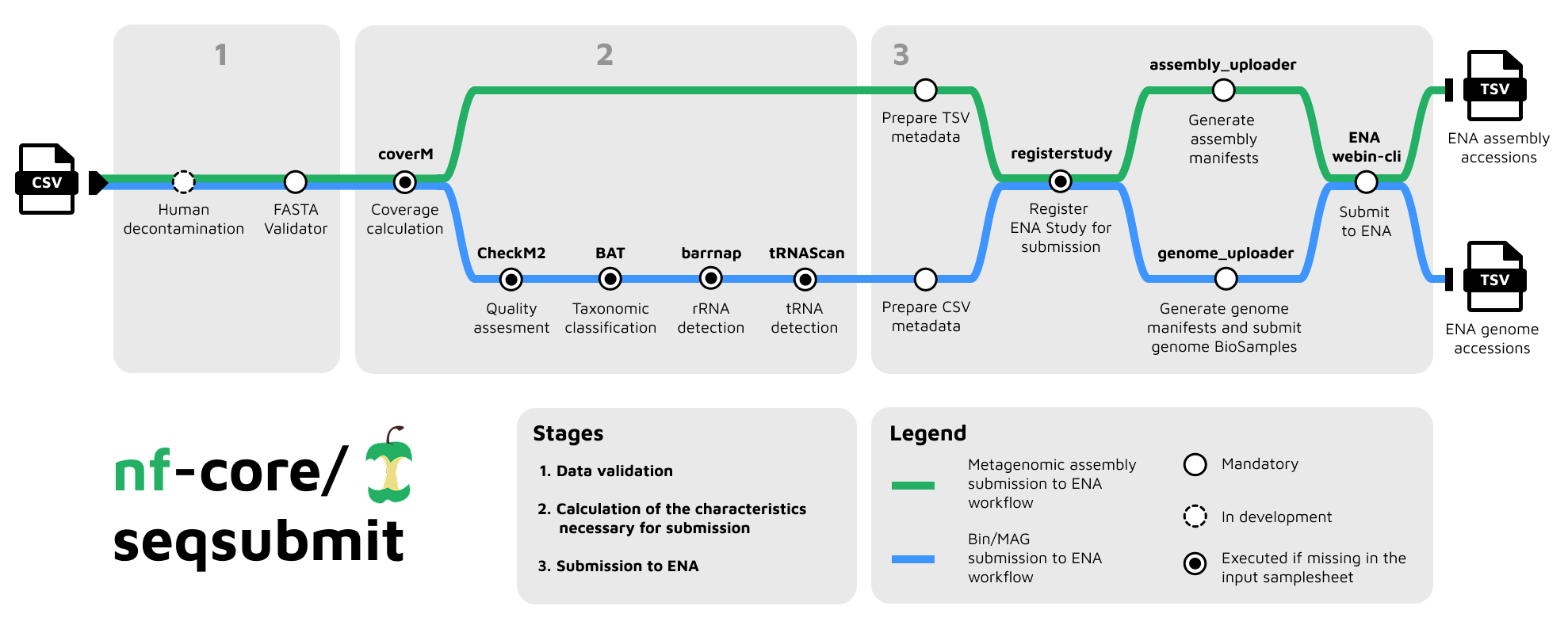
nf-core/seqsubmit is a Nextflow pipeline for submitting sequence data to ENA. Currently, the pipeline supports three submission modes, each routed to a dedicated workflow and requiring its own input samplesheet structure:
magsfor Metagenome Assembled Genomes (MAGs) submission withGENOMESUBMITworkflowbinsfor bins submission withGENOMESUBMITworkflowmetagenomic_assembliesfor assembly submission withASSEMBLYSUBMITworkflow
Requirements
- Nextflow
>=25.04.0 - Webin account registered at https://www.ebi.ac.uk/ena/submit/webin/login
- Raw reads used to assemble contigs submitted to INSDC and associated accessions available
Setup your environment secrets before running the pipeline:
nextflow secrets set ENA_WEBIN "Webin-XXX"
nextflow secrets set ENA_WEBIN_PASSWORD "XXX"
Make sure you update commands above with your authorised credentials.
Input samplesheets
For detailed descriptions of all samplesheet columns, see the usage documentation.
mags and bins modes (GENOMESUBMIT)
The input must follow assets/schema_input_genome.json.
Required columns:
samplefasta(must end with.fa.gz,.fasta.gz, or.fna.gz)accessionassembly_softwarebinning_softwarebinning_parametersmetagenomeenvironmental_mediumbroad_environmentlocal_environmentco-assembly
At least one of the following must be provided per row:
- reads (
fastq_1, optionalfastq_2for paired-end) genome_coverage
Additional supported columns:
stats_generation_softwarecompletenesscontaminationRNA_presenceNCBI_lineage
If genome_coverage, stats_generation_software, completeness, contamination, RNA_presence, or NCBI_lineage are missing, the workflow can calculate or infer them when the required inputs are available.
Those fields are metadata required for the genome_uploader package.
Example samplesheet_genomes.csv:
sample,fasta,accession,fastq_1,fastq_2,assembly_software,binning_software,binning_parameters,stats_generation_software,completeness,contamination,genome_coverage,metagenome,co-assembly,broad_environment,local_environment,environmental_medium,RNA_presence,NCBI_lineage
lachnospira_eligens,data/bin_lachnospira_eligens.fa.gz,SRR24458089,,,spades_v3.15.5,metabat2_v2.6,default,CheckM2_v1.0.1,61.0,0.21,32.07,sediment metagenome,No,marine,cable_bacteria,marine_sediment,No,d__Bacteria;p__Proteobacteria;s__unclassified_ProteobacteriaSamplesheet column requirements: All columns shown in the example above must be present in your samplesheet, even if some values are empty. Columns must be in exactly the same order as shown.
metagenomic_assemblies mode (ASSEMBLYSUBMIT)
The input must follow assets/schema_input_assembly.json.
Required columns:
samplefasta(must end with.fa.gz,.fasta.gz, or.fna.gz)run_accessionassemblerassembler_version
At least one of the following must be provided per row:
- reads (
fastq_1, optionalfastq_2for paired-end) coverage
If coverage is missing and reads are provided, the workflow calculates average coverage with coverm.
Example samplesheet_assembly.csv:
sample,fasta,fastq_1,fastq_2,coverage,run_accession,assembler,assembler_version
assembly_1,data/contigs_1.fasta.gz,data/reads_1.fastq.gz,data/reads_2.fastq.gz,,ERR011322,SPAdes,3.15.5
assembly_2,data/contigs_2.fasta.gz,,,42.7,ERR011323,MEGAHIT,1.2.9Samplesheet column requirements: All columns shown in the example above must be present in your samplesheet, even if some values are empty. Columns must be in exactly the same order as shown.
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Submission study
All data submitted through this pipeline must be associated with an ENA study (project). You can either pass an accession of your existing study via --submission_studyor provide a metadata file via --study_metadata and the pipeline will register the study with ENA before submitting your data.
See the usage documentation for more details.
Database setup (CheckM2 and CAT_pack)
The mags/bins workflow requires databases for completeness/contamination estimation and taxonomy assignment. See Usage documentation for details.
Required parameters:
| Parameter | Description |
|---|---|
--mode | Type of the data to be submitted. Options: [mags, bins, metagenomic_assemblies] |
--input | Path to the samplesheet describing the data to be submitted |
--outdir | Path to the output directory for pipeline results |
--submission_study OR --study_metadata | ENA study accession (PRJ/ERP) to submit the data to OR metadata file in JSON/TSV/CSV format to register new study |
--centre_name | Name of the submitter’s organisation |
Optional parameters:
| Parameter | Description |
|---|---|
--upload_tpa | Flag to control the type of assembly study (third party assembly or not). Default: false |
--test_upload | Upload to TEST ENA server instead of LIVE. Default: true |
--webincli_submit | If set to false, submissions will be validated, but not submitted. Default: true |
General command template:
nextflow run nf-core/seqsubmit \
-profile <docker/singularity/...> \
--mode <mags|bins|metagenomic_assemblies> \
--input <samplesheet.csv> \
--centre_name <your_centre> \
--submission_study <your_study> \
--outdir <outdir>Validation run (submission to the ENA TEST server) in mags mode:
nextflow run nf-core/seqsubmit \
-profile docker \
--mode mags \
--input assets/samplesheet_genomes.csv \
--submission_study <your_study> \
--centre_name TEST_CENTER \
--webincli_submit true \
--test_upload true \
--outdir results/validate_magsValidation run (submission to the ENA TEST server) in metagenomic_assemblies mode:
nextflow run nf-core/seqsubmit \
-profile docker \
--mode metagenomic_assemblies \
--input assets/samplesheet_assembly.csv \
--submission_study <your_study> \
--centre_name TEST_CENTER \
--webincli_submit true \
--test_upload true \
--outdir results/validate_assembliesLive submission example:
nextflow run nf-core/seqsubmit \
-profile docker \
--mode metagenomic_assemblies \
--input assets/samplesheet_assembly.csv \
--submission_study PRJEB98843 \
--test_upload false \
--webincli_submit true \
--outdir results/live_assemblyPlease provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
Key output locations in --outdir:
mags/orbins/: genome metadata, manifests, and per-sample submission support filesmetagenomic_assemblies/: assembly metadata CSVs and per-sample coverage filesmultiqc/: MultiQC summary reportpipeline_info/: execution reports, trace, DAG, and software versions
For full details, see the output documentation.
Credits
nf-core/seqsubmit was originally written by Martin Beracochea, Ekaterina Sakharova, Sofiia Ochkalova, Evangelos Karatzas.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #seqsubmit channel (you can join with this invite).
Citations
If you use this pipeline please make sure to cite all used software. This pipeline uses code and infrastructure developed and maintained by the nf-core community, reused here under the MIT license.
MGnify: the microbiome sequence data analysis resource in 2023
Richardson L, Allen B, Baldi G, Beracochea M, Bileschi ML, Burdett T, et al.
Vol. 51, Nucleic Acids Research. Oxford University Press (OUP); 2022. p. D753–9. Available from: http://dx.doi.org/10.1093/nar/gkac1080
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.