nf-core/funcscan
(Meta-)genome screening for functional and natural product gene sequences
Introduction
The output of nf-core/funcscan provides reports for each of the functional groups:
- antibiotic resistance genes (tools: ABRicate, AMRFinderPlus, DeepARG, fARGene, RGI - summarised by hAMRonization. Results from ABRicate, AMRFinderPlus, and DeepARG are normalised to ARO by argNorm.)
- antimicrobial peptides (tools: Macrel, AMPlify, ampir, hmmsearch - summarised by AMPcombi)
- biosynthetic gene clusters (tools: antiSMASH, DeepBGC, GECCO, hmmsearch, BiGSLiCE - summarised by comBGC)
- carbohydrate-active enzymes (CAZymes), CAZyme gene clusters and substrates (tools: run_dbcan)
As a general workflow, we recommend to first look at the summary reports (ARGs, AMPs, BGCs), to get a general overview of what hits have been found across all the tools of each functional group. After which, you can explore the specific output directories of each tool to get more detailed information about each result. The tool-specific output directories also includes the output from the functional annotation steps of either prokka, pyrodigal, prodigal, or Bakta if the --save_annotations flag was set. Additionally, taxonomic classifications from MMseqs2 are saved if the --taxa_classification_mmseqs_db_savetmp and --taxa_classification_mmseqs_taxonomy_savetmp flags are set.
Similarly, all downloaded databases are saved (i.e. from MMseqs2, antiSMASH, AMRFinderPlus, Bakta, DeepARG, RGI, AMPcombi, and/or run_dbcan) into the output directory <outdir>/databases/ if the --save_db flag was set.
Furthermore, for reproducibility, versions of all software used in the run is presented in a MultiQC report.
The directories listed below will be created in the results directory (specified by the --outdir flag) after the pipeline has finished. All paths are relative to this top-level output directory. The default directory structure of nf-core/funcscan is:
results/├── amp/| ├── ampir/| ├── amplify/| ├── hmmsearch/| └── macrel/├── annotation/| ├── bakta/| ├── prodigal/| ├── prokka/| └── pyrodigal/├── arg/| ├── abricate/| ├── amrfinderplus/| ├── argnorm/| ├── deeparg/| ├── fargene/| ├── hamronization/| └── rgi/├── bgc/| ├── antismash/| ├── bigslice/| ├── deepbgc/| ├── gecco/| └── hmmsearch/├── cazyme/| └── dbcan/├── databases/├── multiqc/├── pipeline_info/├── protein_annotation/| └── interproscan/├── qc/| └── seqkit/├── reports/| ├── ampcombi/| ├── combgc/| └── hamronization_summarize/└── taxonomic_classification/ └── mmseqs_createtsv/work/Pipeline overview
The pipeline is built using Nextflow and processes prokaryotic sequence data through the following steps:
Input contig QC with:
- SeqKit (default) - for separating into long- and short- categories
Taxonomy classification of nucleotide sequences with:
- MMseqs2 (default) - for contig taxonomic classification using 2bLCA.
ORF prediction and annotation with any of:
- Pyrodigal (default) - for open reading frame prediction.
- Prodigal - for open reading frame prediction.
- Prokka - open reading frame prediction and functional protein annotation.
- Bakta - open reading frame prediction and functional protein annotation.
CDS domain annotation:
- InterProScan (default) - for open reading frame protein and domain predictions.
Antimicrobial Resistance Genes (ARGs):
- ABRicate - antimicrobial resistance gene detection, based on alignment to one of several databases.
- AMRFinderPlus - antimicrobial resistance gene detection, using NCBI’s curated Reference Gene Database and curated collection of Hidden Markov Models.
- DeepARG - antimicrobial resistance gene detection, using a deep learning model.
- fARGene - antimicrobial resistance gene detection, using Hidden Markov Models.
- RGI - antimicrobial resistance gene detection, based on alignment to the CARD database.
Antimicrobial Peptides (AMPs):
- ampir - antimicrobial peptide detection, based on a supervised statistical machine learning approach.
- amplify - antimicrobial peptide detection, using a deep learning model.
- hmmsearch - antimicrobial peptide detection, based on hidden Markov models.
- Macrel - antimicrobial peptide detection, using a machine learning approach.
Biosynthetic Gene Clusters (BGCs):
- antiSMASH - biosynthetic gene cluster detection.
- deepBGC - biosynthetic gene cluster detection, using a deep learning model.
- GECCO - biosynthetic gene cluster detection, using Conditional Random Fields (CRFs).
- hmmsearch - biosynthetic gene cluster detection, based on hidden Markov models.
Carbohydrate-active enzymes (CAZYMEs)
- run_dbcan - carbohydrate-active enzyme (CAZyme), CAZyme gene clusters and substrate detection.
Output Summaries:
- AMPcombi - summary report of antimicrobial peptide gene output from various detection tools
- hAMRonization - summary of antimicrobial resistance gene output from various detection tools
- argNorm - Normalize ARG annotations from ABRicate, AMRFinderPlus, and DeepARG to the ARO
- comBGC - summary of biosynthetic gene cluster output from various detection tools
- MultiQC - report of all software and versions used in the pipeline
- Pipeline information - report metrics generated during the workflow execution
Tool details
Taxonomic classification tools
MMseqs2
Output files
taxonomic_classification/mmseqs2_createtsv/<samplename>/:*.tsv: tab-separated table containing the taxonomic lineage of every contig. When a contig cannot be classified according to the database, it is assigned in the ‘lineage’ column as ‘no rank | unclassified’.
reports/<workflow>/<workflow>_complete_summary_taxonomy.tsv.gz: tab-separated table containing the concatenated results from thesummary tables along with the taxonomic classification if the parameter --run_taxa_classificationis called.
MMseqs2 classifies the taxonomic lineage of contigs based on the last common ancestor. The inferred taxonomic lineages are included in the final workflow summaries to annotate the potential source bacteria of the identified AMPs, ARGs, and/or BGCs.
Annotation tools
Pyrodigal, Prodigal, Prokka, Bakta
Prodigal
Output files
prodigal/category/: indicates whether annotation files are of all contigs orlong-only contigs (BGC subworkflow only)<samplename>/:*.fna: nucleotide FASTA file of the input contig sequences*.faa: protein FASTA file of the translated CDS sequences*.gbk: annotation in GBK format, containing both sequences and annotations
Descriptions taken from the Prodigal documentation
Prodigal annotates whole (meta-)genomes by identifying ORFs in a set of genomic DNA sequences. The output is used by some of the functional screening tools.
Pyrodigal
Output files
pyrodigal/category/: indicates whether annotation files are of all contigs orlong-only contigs (BGC subworkflow only)<samplename>/:*.gbk: annotation in GBK format, containing both sequences and annotations*.fna: nucleotide FASTA file of the annotated CDS sequences*.faa: protein FASTA file of the translated CDS sequences
Descriptions taken from the Pyrodigal documentation
Pyrodigal annotates whole (meta-)genomes by identifying ORFs in a set of genomic DNA sequences. It produces the same results as Prodigal while being more resource-optimized, thus faster. Unlike Prodigal, Pyrodigal cannot produce output in GenBank format. The output is used by some of the functional screening tools.
Prokka
Output files
prokka/category/: indicates whether annotation files are of all contigs orlong-only contigs (BGC subworkflow only)<samplename>/*.gff: annotation in GFF3 format, containing both sequences and annotations*.gbk: standard Genbank file derived from the master .gff*.fna: nucleotide FASTA file of the input contig sequences*.faa: protein FASTA file of the translated CDS sequences*.ffn: nucleotide FASTA file of all the prediction transcripts (CDS, rRNA, tRNA, tmRNA, misc_RNA)*.sqn: an ASN1 format “Sequin” file for submission to Genbank*.fsa: nucleotide FASTA file of the input contig sequences, used by “tbl2asn” to create the .sqn file*.tbl: feature Table file, used by “tbl2asn” to create the .sqn file*.err: unacceptable annotations - the NCBI discrepancy report*.log: logging output that Prokka produced during its run*.txt: statistics relating to the annotated features found*.tsv: tab-separated file of all features
Descriptions directly from the Prokka documentation
Prokka performs whole genome annotation to identify features of interest in a set of (meta-)genomic DNA sequences. The output is used by some of the functional screening tools.
Bakta
Output files
bakta/category/: indicates whether annotation files are of all contigs orlong-only contigs (BGC only)<samplename><samplename>.gff3: annotations & sequences in GFF3 format<samplename>.gbff: annotations & sequences in (multi) GenBank format<samplename>.ffn: feature nucleotide sequences as FASTA<samplename>.fna: replicon/contig DNA sequences as FASTA<samplename>.embl: annotations & sequences in (multi) EMBL format<samplename>.faa: CDS/sORF amino acid sequences as FASTA<samplename>_hypothetical.faa: further information on hypothetical protein CDS as simple human readble tab separated values<samplename>_hypothetical.tsv: hypothetical protein CDS amino acid sequences as FASTA<samplename>.tsv: annotations as simple human readble TSV<samplename>.txt: summary in TXT format
Descriptions taken from the Bakta documentation.
Bakta is a tool for the rapid & standardised annotation of bacterial genomes and plasmids from both isolates and MAGs. It provides dbxref-rich, sORF-including and taxon-independent annotations in machine-readable JSON & bioinformatics standard file formats for automated downstream analysis. The output is used by some of the functional screening tools.
Protein annotation
InterProScan
Output files
interproscan/<samplename>_cleaned.faa: clean version of the fasta files (in amino acid format) generated by one of the annotation tools (i.e. Pyrodigal, Prokka, Bakta). These contain sequences with no special characters (for eg.*or-).<samplename>_interproscan_faa.tsv: predicted proteins and domains using the InterPro database in TSV format
InterProScan is designed to predict protein functions and provide possible domain and motif information of the coding regions. It utilizes the InterPro database that consists of multiple sister databases such as PANTHER, ProSite, Pfam, etc. More details can be found in the documentation.
AMP detection tools
ampir, AMPlify, hmmsearch, Macrel
ampir
Output files
ampir/<samplename>.ampir.faa: predicted AMP sequences in FAA format<samplename>.ampir.tsv: predicted AMP metadata in TSV format; contains contig name, sequence and probability score.
ampir (antimicrobial peptide prediction in r) was designed to predict antimicrobial peptides (AMPs) from any given size protein dataset. ampir uses a supervised statistical machine learning approach to predict AMPs. It incorporates two support vector machine classification models, “precursor” and “mature” that have been trained on publicly available antimicrobial peptide data.
AMPlify
Output files
amplify/*_results.tsv: table of contig amino-acid sequences with prediction result (AMP or non-AMP) and information on sequence length, charge, probability score, AMPlify log-scaled score
AMPlify is an attentive deep learning model for antimicrobial peptide prediction. It takes in contig annotations (as protein sequences) and classifies them as either AMP or non-AMP.
hmmsearch
Output files
hmmersearch/*.txt.gz: human readable output summarizing hmmsearch results*.sto.gz: optional multiple sequence alignment (MSA) in Stockholm format*.tbl.gz: optional tabular (space-delimited) summary of per-target output*.domtbl.gz: optional tabular (space-delimited) summary of per-domain output
HMMER/hmmsearch is used for searching sequence databases for sequence homologs, and for making sequence alignments. It implements methods using probabilistic models called profile hidden Markov models (profile HMMs). hmmsearch is used to search one or more profiles against a sequence database.
Macrel
Output files
macrel_contigs/*.smorfs.faa.gz: zipped fasta file containing amino acid sequences of small peptides (<100 aa, small open reading frames) showing the general gene prediction information in the contigs*.all_orfs.faa.gz: zipped fasta file containing amino acid sequences showing the general gene prediction information in the contigsprediction.gz: zipped file, with all predicted and non-predicted AMPs in a table format*.md: readme file containing tool specific information (e.g. citations, details about the output, etc.)*_log.txt: log file containing the information pertaining to the run
Macrel is a tool that mines antimicrobial peptides (AMPs) from (meta)genomes by predicting peptides from genomes (provided as contigs) and outputs predicted antimicrobial peptides that meet specific criteria/thresholds.
ARG detection tools
ABRicate, AMRFinderPlus, DeepARG, fARGene, RGI.
ABRicate
Output files
abricate/*.{csv,tsv}: search results in tabular format
ABRicate screens contigs for antimicrobial resistance or virulence genes. It comes bundled with multiple databases: NCBI, CARD, ARG-ANNOT, Resfinder, MEGARES, EcOH, PlasmidFinder, Ecoli_VF and VFDB.
AMRFinderPlus
Output files
amrfinderplus/*.tsv: search results in tabular format
AMRFinderPlus relies on NCBI’s curated Reference Gene Database and curated collection of Hidden Markov Models. It identifies antimicrobial resistance genes, resistance-associated point mutations, and select other classes of genes using protein annotations and/or assembled nucleotide sequences.
DeepARG
Output files
deeparg/*.align.daa*: Intermediate DIAMOND alignment output*.align.daa.tsv: DIAMOND alignment output as .tsv*.mapping.ARG: ARG predictions with a probability >= –prob (0.8 default).*.mapping.potential.ARG: ARG predictions with a probability < –prob (0.8 default)
deepARG uses deep learning to characterize and annotate antibiotic resistance genes in metagenomes. It is composed of two models for two types of input: short sequence reads and gene-like sequences. In this pipeline we use the ls model, which is suitable for annotating full sequence genes and to discover novel antibiotic resistance genes from assembled samples. The tool DIAMOND is used as an aligner.
fARGene
Output files
fargene/fargene_analysis.log: logging output that fARGene produced during its run<sample_name>/:hmmsearchresults/: output from intermediate hmmsearch steppredictedGenes/:*-filtered.fasta: nucleotide sequences of predicted ARGs*-filtered-peptides.fasta: amino acid sequences of predicted ARGs
results_summary.txt: text summary of results, listing predicted genes and ORFs for each input filetmpdir/: temporary output files and fasta files (only if--arg_fargene_savetmpfilessupplied)
fARGene (Fragmented Antibiotic Resistance Gene Identifier) is a tool that takes either fragmented metagenomic data or longer sequences as input and predicts and delivers full-length antibiotic resistance genes as output. The tool includes developed and optimised models for a number of resistance gene types. By default the pipeline runs all models, thus you will receive output for all models. If only a sub-list or single model is required, this can be specified with the --hmm-model flag. Available models are:
class_a: class A beta-lactamasesclass_b_1_2: subclass B1 and B2 beta-lactamasesclass_b3: subclass B3 beta-lactamasesclass_c: class C beta-lactamasesclass_d_1, class_d_2: class D beta-lactamasesqnr: quinolone resistance genestet_efflux,tet_rpg,tet_enzyme: tetracycline resistance genes
RGI
Output files
rgi/<samplename>.txt: hit results table separated by ‘#’<samplename>.json: hit results in json format (only if--arg_rgi_savejsonsupplied)temp/:<samplename>.fasta.temp.*.json: temporary json files, ‘*’ stands for ‘homolog’, ‘overexpression’, ‘predictedGenes’ and ‘predictedGenes.protein’ (only if--arg_rgi_savetmpfilessupplied).
RGI (Resistance Gene Identifier) predicts resistome(s) from protein or nucleotide data based on homology and SNP models. It uses reference data from the Comprehensive Antibiotic Resistance Database (CARD).
BGC detection tools
antiSMASH, deepBGC, GECCO, hmmsearch, BiGSLiCE.
Note that the BGC tools are run on a set of annotations generated on only long contigs (3000 bp or longer) by default. These specific filtered FASTA files are under bgc/seqkit/, and annotations files are under annotation/<annotation_tool>/long/, if the corresponding saving flags are specified (see parameter docs). However the same annotations should also be annotation files in the sister all/ directory.
Input contig QC
Output files
seqkit/<samplename>_long.fasta: FASTA file containing contigs equal or longer than the threshold set by--contig_qc_lengththresholdused in BGC subworkflow
SeqKit is a cross-platform and ultrafast toolkit for FASTA/Q file manipulation.
Note that filtered FASTA is only used for BGC workflow for run-time optimisation and biological reasons. All contigs are otherwise screened in ARG/AMP workflows.
antiSMASH
Output files
antismash/css: accessory files for the HTML outputclusterblastoutput.txt(optional): raw BLAST output of known clusters previously predicted by antiSMASH using the built-in ClusterBlast algorithmimages: accessory files for the HTML outputindex.html: interactive web view of results in HTML formatjs: accessory files for the HTML outputknownclusterblast/: directory with MIBiG hits (optional)*_c*.txt: tables with MIBiG hits
knownclusterblastoutput.txt(optional): raw BLAST output of known clusters of the MIBiG database.regions.js: sideloaded annotations of protoclusters and/or subregions*region*.gbk: nucleotide sequence + annotations in GenBank file format; one file per antiSMASH hit.<sample name>.gbk: nucleotide sequence and annotations in GenBank format; converted from input file<sample name>.json: nucleotide sequence and annotations in JSON format; converted from GenBank file<sample name>.log: logging output that antiSMASH produced during its run<sample name>.zip: compressed version of the output folder in zip format
antiSMASH (antibiotics & Secondary Metabolite Analysis SHell) is a tool for rapid genome-wide identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial genomes. It identifies biosynthetic loci covering all currently known secondary metabolite compound classes in a rule-based fashion using profile HMMs and aligns the identified regions at the gene cluster level to their nearest relatives from a database containing experimentally verified gene clusters (MIBiG).
deepBGC
Output files
deepbgc/README.txt: Summary of output files generatedLOG.txt: Log output of DeepBGC*.antismash.json: AntiSMASH JSON file for sideloading*.bgc.gbk: Sequences and features of all detected BGCs in GenBank format*.bgc.tsv: Table of detected BGCs and their properties*.full.gbk: Fully annotated input sequence with proteins, Pfam domains (PFAM_domain features) and BGCs (cluster features)*.pfam.tsv: Table of Pfam domains (pfam_id) from given sequence (sequence_id) in genomic order, with BGC detection scoresevaluation/*.bgc.png: Detected BGCs plotted by their nucleotide coordinates*.pr.png: Precision-Recall curve based on predicted per-Pfam BGC scores*.roc.png: ROC curve based on predicted per-Pfam BGC scores*.score.png: BGC detection scores of each Pfam domain in genomic order
deepBGC detects BGCs in bacterial and fungal genomes using deep learning. DeepBGC employs a Bidirectional Long Short-Term Memory Recurrent Neural Network and a word2vec-like vector embedding of Pfam protein domains. Product class and activity of detected BGCs is predicted using a Random Forest classifier.
GECCO
Output files
gecco/*.genes.tsv/: TSV file containing detected/predicted genes with BGC probability scores*.features.tsv: TSV file containing identified domains*.clusters.tsv: TSV file containing coordinates of predicted clusters and BGC types*_cluster_*.gbk: GenBank file (if clusters were found) containing sequence with annotations; one file per GECCO hit*.gff: GFF3 converted cluster tables containing the position and metadata for all the predicted clusters (only if--bgc_gecco_runconvert --bgc_gecco_convertmode clusters --bgc_gecco_convertformat gff)*.region*.gbk: Converted and aliased GenBank files so that they can be loaded by BiG-SLiCE (only if--bgc_gecco_runconvert --bgc_gecco_convertmode gbk --bgc_gecco_convertformat bigslice)*.faa: Amino-acid FASTA converted GenBank files of all the proteins in a cluster (only if--bgc_gecco_runconvert --bgc_gecco_convertmode gbk --bgc_gecco_convertformat faa)*.fnasequence FASTA converted GenBank files from the cluster (only if--bgc_gecco_runconvert --bgc_gecco_convertmode gbk --bgc_gecco_convertformat fna)
GECCO is a fast and scalable method for identifying putative novel Biosynthetic Gene Clusters (BGCs) in genomic and metagenomic data using Conditional Random Fields (CRFs).
The additional GFF3, GenBank, or FASTA files from --bgc_gecco_runconvert, can be useful for additional further analysis of the BGC hits.
BiGSLiCE
Output files
bigslice/<samplename>/result/data.db: SQLite database containing results for BGCs, CDSs, Gene Cluster Families (GCFs), HMMs and HSPs.tsv_export/(optional): TSV exports of all parsed BGC metadata, vectorized features and clustering results. Only produced when--bgc_bigslice_exporttsvis set.tmp/<run_id>/*.fa: predicted biosynthetic features as FASTA files, one file per hit HMM.
BiG-SLiCE (Biosynthetic Gene cluster Super-Linear Clustering Engine) is a highly scalable tool for the large-scale analysis and clustering of Biosynthetic Gene Clusters (BGCs) into Gene Cluster Families (GCFs).
It takes BGC regions in GenBank format (e.g. output from antiSMASH or GECCO) along with an HMM database and produces an SQLite database of predicted BGC features and GCF assignments.
BiG-SLiCE requires the HMM database to be supplied via --bgc_bigslice_db and is activated with --bgc_run_bigslice. It requires at least one of antiSMASH or GECCO (with convert in bigslice format) to be enabled.
All results are stored in a SQLite database (data.db) which can be explored with standard SQLite tools or via the BiG-SLiCE interactive web interface.
CAZyme annotation tools
run_dbcan
Output files
cazyme/dbcan/cazyme_annotation/<sample.id>_overview.tsv: TSV file containing the results of dbCAN CAZyme annotation<sample.id>_dbCAN_hmm_results.tsv: TSV file containing the detailed dbCAN HMM results for CAZyme annotation<sample.id>_dbCANsub_hmm_results.tsv: TSV file containing the detailed dbCAN subfamily results for CAZyme annotation<sample.id>_diamond.out: TSV file containing the detailed dbCAN diamond results for CAZyme annotation
cgc/<sample.id>_cgc.gff: GFF file containing the CAZyme gene clusters (CGC) identified by dbCAN. This file is generated from the dbCAN annotation and contains the locations of CAZyme gene clusters in the genome<sample.id>_cgc_standard_out.tsv: Standard output file from dbCAN for CAZyme gene clusters (CGC) in a tabular format. This file summarizes the CAZyme gene clusters identified in the genome<sample.id>_diamond.out.tc: TSV file containing the diamond output for transporter annotation<sample.id>_TF_hmm_results.tsv: TSV file containing the results of transcription factor screening<sample.id>_STP_hmm_results.tsv: TSV file containing the results of signaling transduction proteins (STP) annotation
substrate/<sample.id>_total_cgc_info.tsv: TSV file summarizing the total additional genes in the genome<sample.id>_substrate_prediction.tsv: TSV file containing the substrate predictions based on the CGC annotations from dbCAN<sample.id>_synteny_pdf/: Directory containing one or more PDF files showing the syntenic regions of the CGCs in DNA sequence as identified by dbCAN
run_dbcan is an automated tool for carbohydrate-active enzyme (CAZyme), CAZyme gene cluster and substrate annotation.
Summary tools
AMPcombi, hAMRonization, comBGC, MultiQC, pipeline information, argNorm.
AMPcombi
Output files
ampcombi/Ampcombi_summary.tsv: tab-separated table containing the concatenated and filtered results from each AMPcombi summary table. This is the output given when the taxonomic classification is not activated (pipeline default).Ampcombi_parse_tables.log: log file containing the run information from AMPcombi submoduleampcombi2/parsetablesAmpcombi_complete.log: log file containing the run information from AMPcombi submoduleampcombi2/completeAmpcombi_summary_cluster.tsv: tab-separated table containing the clustered AMP hits. This is the output given when the taxonomic classification is not activated (pipeline default).Ampcombi_summary_cluster_representative_seq.tsv: tab-separated table containing the representative sequence of each cluster. This can be used in AMPcombi for constructing 3D structures using ColabFold. For more details on how to do this, please refer to the AMPcombi documentation.Ampcombi_cluster.log: log file containing the run information from AMPcombi submoduleampcombi2/clusterampcombi_complete_summary_taxonomy.tsv.gz: summarised output from all AMP workflow tools with taxonomic assignment in compressed tsv format. This is the same output asAmpcombi_summary_cluster.tsvfile but with taxonomic classification of the contig.<sample>/contig_gbks: contains all the contigs in gbk format that an AMP was found on using the custom parameters<sample>/*_ampcombi.log: a log file generated by AMPcombi<sample>/*_ampcombi.tsv: summarised output in tsv format for each sample<sample>/*_amp.faa*: fasta file containing the amino acid sequences for all AMP hits for each sample<sample>/*_mmseqs_matches.txt*: alignment file generated by MMseqs2 for each sample
In some cases when the AMP and the taxonomic classification subworkflows are turned on, it can happen that only summary files per sample are created in the output folder with no Ampcombi_summary.tsv and Ampcombi_summary_cluster.tsv files with no taxonomic classifications merged.
This can occur if some AMP prediction parameters are ‘too strict’ or only one AMP tool is run, which can lead to no AMP hits found in any of the samples or in only one sample.
Look out for the warning [nf-core/funcscan] AMPCOMBI2: 0/1 file passed. Skipping AMPCOMBI2_COMPLETE, AMPCOMBI2_CLUSTER, and TAXONOMY MERGING steps. in the stdout or .nextflow.log file.
In that case we recommend to lower the AMP prediction thresholds and run more than one AMP prediction tool.
AMP summary table header descriptions using DRAMP as reference database
| Table column | Description |
|---|---|
name |
Name of the sample |
contig_id |
Contig header |
prob_macrel |
Probability associated with the AMP prediction using MACREL |
prob_neubi |
Probability associated with the AMP prediction using NEUBI |
prob_ampir |
Probability associated with the AMP prediction using AMPIR |
prob_amplify |
Probability associated with the AMP prediction using AMPLIFY |
evalue_hmmer |
Expected number of false positives (nonhomologous sequences) with a similar of higher score. This stands for how significant the hit is, the lower the evalue, the more significant the hit |
aa_sequence |
Amino acid sequence that forms part of the contig and is AMP encoding |
target_id |
DRAMP ID within the database found to be similar to the predicted AMP by MMseqs2 alignment |
pident |
Percentage identity of amino acid residues that fully aligned between the DRAMP sequence and the predicted AMP sequence |
evalue |
Number of alignments of similar or better qualities that can be expected when searching a database of similar size with a random sequence distribution. This is generated by MMseqs2 alignments using the DRAMP AMP database. The lower the value the more significant that the hit is positive. An e-value of < 0.001 means that the this hit will be found by chance once per 1,0000 queries |
Sequence |
Sequence corresponding to the DRAMP ID found to be similar to the predicted AMP sequence |
Sequence_length |
Number of amino acid residues in the DRAMP sequence |
Name |
Full name of the peptide copied from the database it was uploaded to |
Swiss_Prot_Entry |
Entry name of the peptide within the UniProtKB/Swiss-Prot database |
Family |
Name of the family, group or class of AMPs this peptide belongs to, e.g. bacteriocins |
Gene |
Name of the gene (if available in the database) that encodes the peptide |
Source |
Name of the source organism (if available in the database) from which the peptide was extracted |
Activity |
Peptide activity, e.g. Antimicrobial, Antibacterial, Anti-Gram+, Anti-Gram-, Insecticidal or Antifungal |
Protein_existence |
Peptide status, e.g. only a homology, protein level, predicted or transcript level |
Structure |
Type of peptide structure, e.g. alpha helix, bridge, etc. |
Structure_Description |
Further description of the structure if available |
PDB_ID |
The ID of an equivalent peptide found in the protein data bank PDB |
Comments |
Further details found in the database regarding the peptide |
Target_Organism |
Name of the target organism to which the peptide is effective against |
Hemolytic_activity |
Type of hemolytic activity if any |
Linear/Cyclic/Branched |
Whether the hit is a linear, cyclic or branched peptide |
N-terminal_Modification |
Whether it contains N-terminal_Modification |
C-termina_Modification |
Whether it contains C-terminal_Modification |
Other_Modifications |
Whether there are any other modifications found in the peptide structure |
Stereochemistry |
Type of peptide stereochemistry if available |
Cytotoxicity |
Cytotoxicity mechanism of the peptide if available |
Binding_Target |
Peptide binding target, e.g. lipid, cell membrane or chitin binding |
Pubmed_ID |
Pubmed ID if a publication is associated with the peptide |
Reference |
Citation of the associated publication if available |
Author |
Authors’ names associated with the publication or who have uploaded the peptide |
Title |
Publication title if available |
... |
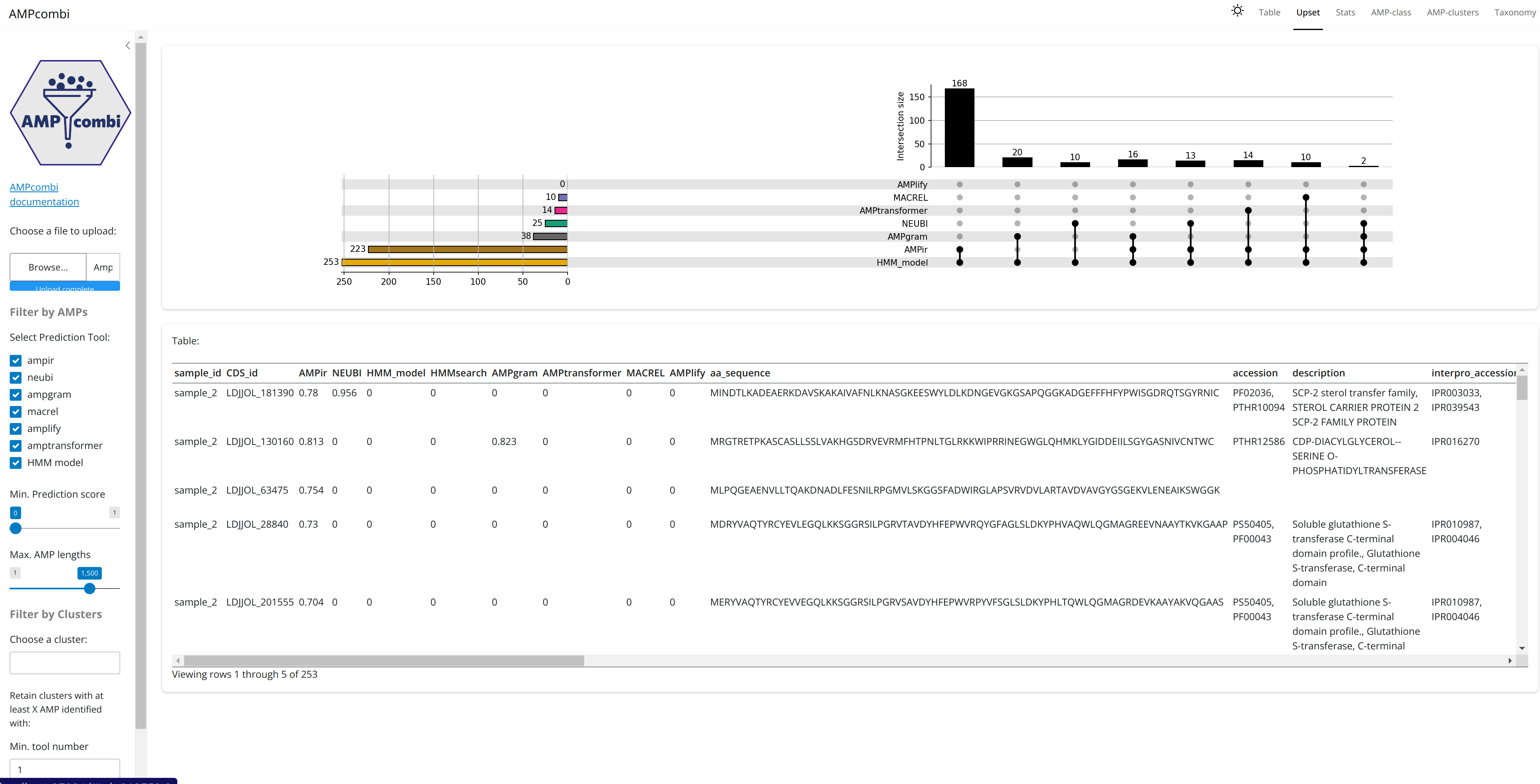
AMPcombi summarizes the results of antimicrobial peptide (AMP) prediction tools (ampir, AMPlify, Macrel, and other non-nf-core supported tools) into a single table and aligns the hits against a reference AMP database for functional, structural and taxonomic classification using MMseqs2.
It further assigns the physiochemical properties (e.g. hydrophobicity, molecular weight) using the Biopython toolkit and clusters the resulting AMP hits from all samples using MMseqs2.
To further filter the recovered AMPs using the presence of signaling peptides, the output file Ampcombi_summary_cluster.tsv or ampcombi_complete_summary_taxonomy.tsv.gz can be used downstream as detailed here.
The final tables generated may also be visualized and explored using an interactive user interface.
hAMRonization
Output files
hamronization_summarize/one of the following:hamronization_combined_report.json: summarised output in .json formathamronization_combined_report.tsv: summarised output in .tsv format when the taxonomic classification is turned off (pipeline default).hamronization_complete_summary_taxonomy.tsv.gz: summarised output in gzipped format when the taxonomic classification is turned on by--run_taxa_classification.hamronization_combined_report.html: interactive output in .html format
ARG summary table headers
| Table column | Description |
|---|---|
input_file_name |
Name of the file containing the sequence data to be analysed |
gene_symbol |
Short name of a gene; a single word that does not contain white space characters. It is typically derived from the gene name |
gene_name |
Name of a gene |
reference_database_name |
Identifier of a biological or bioinformatics database |
reference_database_version |
Version of the database containing the reference sequences used for analysis |
reference_accession |
Identifier that specifies an individual sequence record in a public sequence repository |
analysis_software_name |
Name of a computer package, application, method or function used for the analysis of data |
analysis_software_version |
Version of software used to analyze data |
genetic_variation_type |
Class of genetic variation detected |
antimicrobial_agent (optional) |
A substance that kills or slows the growth of microorganisms, including bacteria, viruses, fungi and protozoans |
coverage_percentage (optional) |
Percentage of the reference sequence covered by the sequence of interest |
coverage_depth (optional) |
Average number of reads representing a given nucleotide in the reconstructed sequence |
coverage_ratio (optional) |
Ratio of the reference sequence covered by the sequence of interest. |
drug_class (optional) |
Set of antibiotic molecules, with similar chemical structures, molecular targets, and/or modes and mechanisms of action |
input_gene_length (optional) |
Length (number of positions) of a target gene sequence submitted by a user |
input_gene_start (optional) |
Position of the first nucleotide in a gene sequence being analysed (input gene sequence) |
input_gene_stop (optional) |
Position of the last nucleotide in a gene sequence being analysed (input gene sequence) |
input_protein_length (optional) |
Length (number of positions) of a protein target sequence submitted by a user |
input_protein_start (optional) |
Position of the first amino acid in a protein sequence being analysed (input protein sequence) |
input_protein_stop (optional) |
Position of the last amino acid in a protein sequence being analysed (input protein sequence) |
input_sequence_id (optional) |
Identifier of molecular sequence(s) or entries from a molecular sequence database |
nucleotide_mutation (optional) |
Nucleotide sequence change(s) detected in the sequence being analysed compared to a reference |
nucleotide_mutation_interpretation (optional) |
Description of identified nucleotide mutation(s) that facilitate clinical interpretation |
predicted_phenotype (optional) |
Characteristic of an organism that is predicted rather than directly measured or observed |
predicted_phenotype_confidence_level (optional) |
Confidence level in a predicted phenotype |
amino_acid_mutation (optional) |
Amino acid sequence change(s) detected in the sequence being analysed compared to a reference |
amino_acid_mutation_interpretation (optional) |
Description of identified amino acid mutation(s) that facilitate clinical interpretation. |
reference_gene_length (optional) |
Length (number of positions) of a gene reference sequence retrieved from a database |
reference_gene_start (optional) |
Position of the first nucleotide in a reference gene sequence |
reference_gene_stop (optional) |
Position of the last nucleotide in a reference sequence |
reference_protein_length (optional) |
Length (number of positions) of a protein reference sequence retrieved from a database |
reference_protein_start (optional) |
Position of the first amino acid in a reference protein sequence |
reference_protein_stop (optional) |
Position of the last amino acid in a reference protein sequence |
resistance_mechanism (optional) |
Antibiotic resistance mechanisms evolve naturally via natural selection through random mutation, but it could also be engineered by applying an evolutionary stress on a population |
strand_orientation (optional) |
Orientation of a genomic element on the double-stranded molecule |
sequence_identity (optional) |
Sequence identity is the number (%) of matches (identical characters) in positions from an alignment of two molecular sequences |
hAMRonization summarizes the outputs of the antimicrobial resistance gene detection tools (ABRicate, AMRFinderPlus, DeepARG, fARGene, RGI) into a single unified tabular format. It supports a variety of summary options including an interactive summary.
argNorm
Output files
normalized/*.{tsv}: search results in tabular format
ARG summary table headers
| Table column | Description |
|---|---|
ARO |
ARO accessions of ARG |
confers_resistance_to |
ARO accessions of drugs to which ARGs confer resistance to |
resistance_to_drug_classes |
ARO accessions of drugs classes to which drugs in confers_resistance_to belong |
argnorm is a tool to normalize antibiotic resistance genes (ARGs) by mapping them to the antibiotic resistance ontology (ARO) created by the CARD database. argNorm also enhances antibiotic resistance gene annotations by providing categorization of the drugs that antibiotic resistance genes confer resistance to.
argNorm takes the outputs of the hAMRonization tool of ABRicate, AMRFinderPlus, and DeepARG and normalizes ARGs in the hAMRonization output to the ARO.
comBGC
Output files
comBGC/combgc_complete_summary.tsv: summarised output from all BGC detection tools used in tsv format (all samples concatenated). This is the output given when the taxonomic classification is not activated (pipeline default).combgc_complete_summary.tsv.gz: summarised output in gzipped format from all BGC detection tools used in tsv format (all samples concatenated) along with the taxonomic classification obtained when--run_taxa_classificationis activated.*/combgc_summary.tsv: summarised output from all applied BGC detection tools in tsv format for each sample.
BGC summary table headers
| Table column | Description |
|---|---|
Sample_ID |
ID of the sample |
Prediction_tool |
BGC prediction tool (antiSMASH, DeepBGC, and/or GECCO) |
Contig_ID |
ID of the contig containing the candidate BGC |
Product_class |
Predicted BGC type/class |
BGC_probability |
Confidence of BGC candidate as inferred by the respective tool |
BGC_complete |
Whether BGC sequence is assumed to be complete or truncated by the edge of the contig |
BGC_start |
Predicted BGC start position on the ontig |
BGC_end |
Predicted BGC end position on the contig |
BGC_length |
Length of the predicted BGC |
CDS_ID |
ID of the coding sequence(s) (CDS) from the annotation step (prodigal/prokka/bakta) if provided by the tool |
CDS_count |
Number of CDSs the BGC contains |
PFAM_domains |
Inferred PFAM IDs or annotations if provided by the tool |
MIBiG_ID |
Inferred MIBiG IDs if provided by the tool |
InterPro_ID |
Inferred InterPro IDs if provided by the tool |
comBGC is a tool built for nf-core/funcscan which summarizes the results of the Biosynthetic Gene Cluster (BGC) prediction tools (antiSMASH, deepBGC, GECCO) used in the pipeline into one comprehensive tabular summary with standardised headers.
ℹ️ comBGC does not feature
hmmer_hmmsearchsupport. Please check the hmmsearch results directory.
MultiQC
Output files
multiqc/multiqc_report.html: a standalone HTML file that can be viewed in your web browsermultiqc_data/: directory containing raw parsed data used for MultiQC report renderingmultiqc_plots/: directory containing any static images from the report in various formats
MultiQC is used in nf-core/funcscan to report the versions of all software used in the given pipeline run, and provides a suggested methods text. This allows for reproducible analysis and transparency in method reporting in publications.
Results generated by MultiQC collate pipeline QC from supported tools. The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability. For more information about how to use MultiQC reports, see http://multiqc.info.
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter’s are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv. - Parameters used by the pipeline run:
params.json.
- Reports generated by Nextflow:
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.