nf-core/genephylomodeler
A bioinformatics pipeline that fits evolutionary models and performs hypothesis testing on multiple sequence alignments
Introduction
nf-core/genephylomodeler is a bioinformatics pipeline that fits evolutionary models and performs hypothesis testing on multiple sequence alignments of coding genes. These include (but are not limited to) detecting signatures of selection and estimating evolutionary rates using methods from HyPhy, PAML, and other packages. The pipeline takes a samplesheet with alignment files and phylogenetic trees as input, applies one or more tools of interest, and produces an output in JSON and text format.
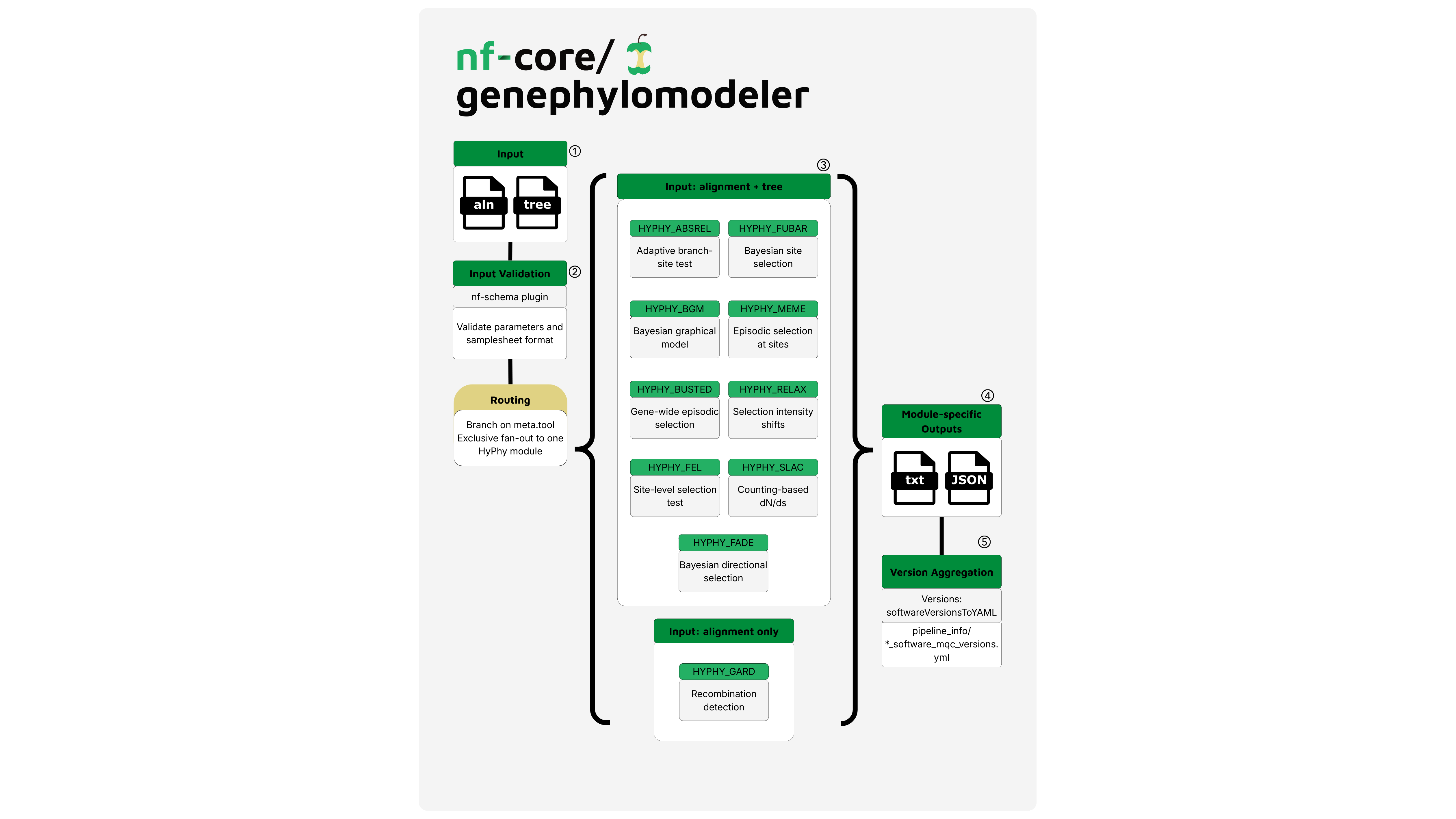
The pipeline currently supports the following tools from HyPhy:
- Adaptive Branch-Site Random Effects Likelihood (
aBSREL) - Bayesian Graphical Model (
BGM) - Branch-Site Unrestricted Statistical Test for Episodic Diversification (
BUSTED) - FUBAR Aproach to Directional Evolution (
FADE) - Fixed Effects Likelihood (
FEL) - Fast, Unconstrained Bayesian AppRoximation (
FUBAR) - Genetic Algorithm for Recombination Detection (
GARD) - Mixed Effects Model of Evolution (
MEME) - Test for Relaxation or Intensification of Selection (
RELAX) - Single-Likelihood Ancestor Counting (
SLAC)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
gene_name,alignment,tree,suite,tool
KSR2,/path/to/ksr2.fna,/path/to/ksr2.tree,hyphy,absrel
KSR2,/path/to/ksr2.fna,/path/to/ksr2.tree,hyphy,busted
TRIM5,/path/to/trim5.fna,/path/to/trim5.tree,hyphy,meme
MX1,/path/to/mx1.fna,/path/to/mx1_labeled.tree,hyphy,relaxEach row represents a multiple sequence alignment for a gene.
Now, you can run the pipeline using:
nextflow run nf-core/genephylomodeler \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/genephylomodeler was originally written by Nina Xiong.
We thank the following people for their extensive assistance in the development of this pipeline:
- Areeba Rahu
- Nuha Kadak
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #genephylomodeler channel (you can join with this invite).
Citations
This pipeline uses methods from the HyPhy (Hypothesis Testing using Phylogenies) suite.
The test data uses an alignment of primate sequences for the KSR2 gene, a kinase suppressor of RAS-2, from Enard et al, 2016.
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.