nf-core/marsseq
MARS-seq v2 pre-processing pipeline with velocity
1.0.0). The latest
stable release is
1.0.3
.
Introduction
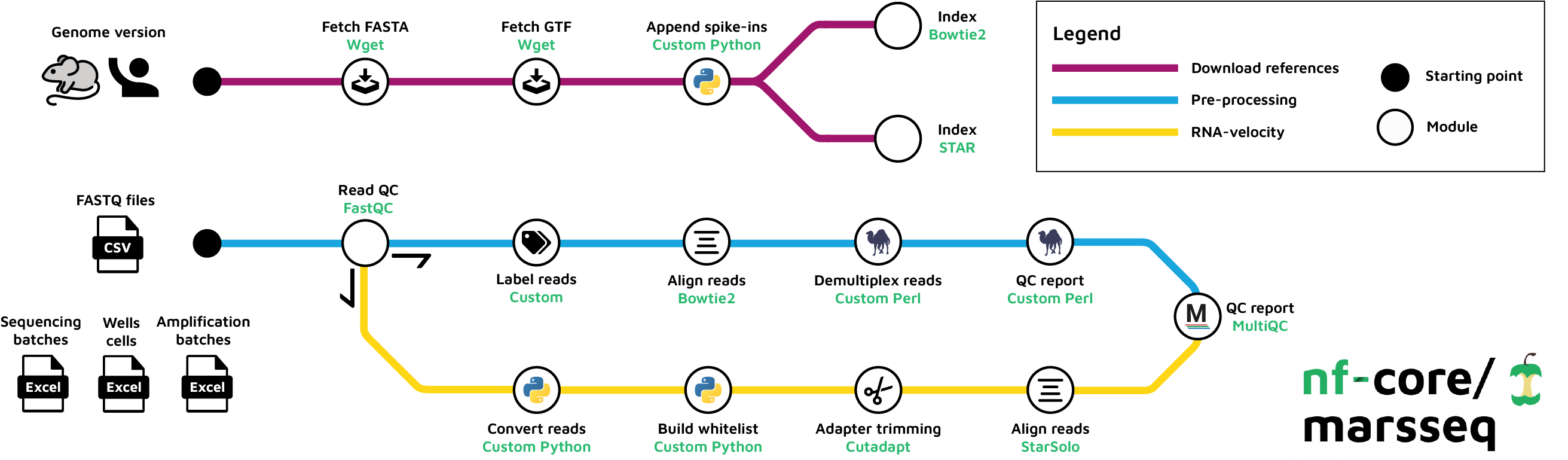
nf-core/marsseq is a bioinformatics pipeline for MARS-seq v2.0 preprocessing pipeline. As an additional work we have developed custom set of scripts to run velocity inference using StarSolo.
We do so by converting the raw reads into 10X v2 format.
Usage
Note If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with
-profile testbefore running the workflow on actual data.
To run the pipeline you have create experiment metadata files:
and samplesheet (samplesheet.csv). We provide test example here.
Next, you have to generate genome references to incorporate ERCC spike-ins. References are downloaded from GENCODE database.
nextflow run nf-core/marsseq \
-profile <docker/singularity/.../institute> \
--genome <mm10,mm9,GRCh38> \
--build_references \
--input samplsheet.csv \
--outdir <OUTDIR>Now, you can run the pipeline using:
nextflow run nf-core/marsseq \
-profile <docker/singularity/.../institute> \
--genome <mm10,mm9,GRCh38> \
--input samplesheet.csv \
--outdir <OUTDIR>Warning: Please provide pipeline parameters via the CLI or Nextflow
-params-fileoption. Custom config files including those provided by the-cNextflow option can be used to provide any configuration except for parameters; see docs.
For more details, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the the results of a test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/marsseq was originally written by Martin Proks.
We thank the following people for their extensive assistance in the development of this pipeline:
- Jose Alejandro Romero Herrera (@joseale2310)
- Maxime Garcia (@maxulysse)
Keren-Shaul, H., Kenigsberg, E., Jaitin, D.A. et al. MARS-seq2.0: an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat Protoc 14, 1841–1862 (2019). https://doi.org/10.1038/s41596-019-0164-4
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #marsseq channel (you can join with this invite).
Citations
If you use nf-core/marsseq for your analysis, please cite it using the following doi: 10.5281/zenodo.8016373
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.