nf-core/metaboigniter
Pre-processing of mass spectrometry-based metabolomics data with quantification and identification based on MS1 and MS2 data.
1.0.1). The latest
stable release is
2.0.1
.
Introduction
nf-core/metaboigniter is bioinformatics pipeline for pre-processing of mass spectrometry-based metabolomics data.
The workflow performs MS1 based quantification and MS2 based identification using combination of different modules. The following steps can be performed using the workflow:
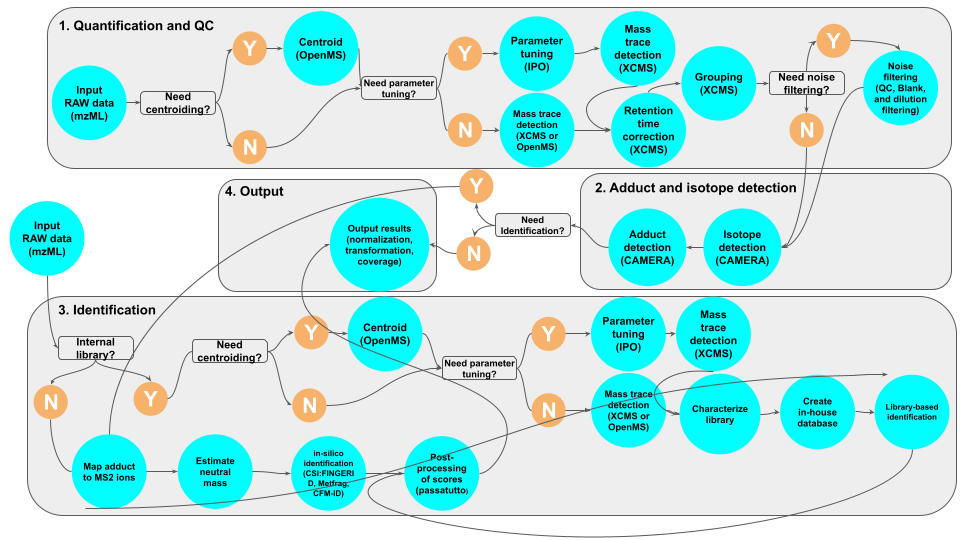
- Centroiding (optional): Also referred to as peak pickering is a step that reduce the distribution of ions derived from a single mass to the peak of the distribution.
- parameter tuning using IPO: MS1 quantification and library characterization parameters can be tuned using IPO
- mass trace detection: The ions derived from the same analytes are clustered together forming a mass trace. These are the entities that will be used for quantification.
- mass trace matching and retention time (RT) correction: The mass traces across different samples will be match against each other and RT shift between the samples will be adjusted.
- filtering (optional): The mass traces will be filtered out based on the QC samples.
- Annotation: The isotopes and adducts will be annotated in this step.
- MS2 (identification): At the moment we support two types of identification: in-silico and identificaiton based on internal library. This will be expanded in the corresponding section. At the moment we do not support MS1-based identification.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible.
Quick Start
-
Install
nextflow(>=20.04.0) -
Install any of
Docker,Singularity,Podman,ShifterorCharliecloudfor full pipeline reproducibility (please only useCondaas a last resort; see docs) -
Download the pipeline and test it on a minimal dataset with a single command
nextflow run nf-core/metaboigniter -profile test,<docker/singularity/podman/shifter/charliecloud/conda/institute>Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment. -
Start running your own analysis!
We highly recommend that you follow our usage documentation. Since the number of parameters is large, it’s going to be a fairly complex bash command to run the workflow. Nevertheless, the parameters can always be passed to the workflow as argument using two dashes ”—”.
See usage docs for all of the available options when running the pipeline.
Documentation
The nf-core/metaboigniter pipeline comes with documentation about the pipeline: usage and output.
Credits
nf-core/metaboigniter was originally written by Payam Emami.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on Slack (you can join with this invite).
Citation
If you use nf-core/metaboigniter for your analysis, please cite it using the following doi: 10.5281/zenodo.4743790
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
In addition, references of tools and data used in this pipeline are as follows:
- Kuhl C, Tautenhahn R, Boettcher C, Larson TR and Neumann S: CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal Chem 84:283-289.
- Benton HP, Wong DM, Trauger SA, Siuzdak G: XCMS2: Processing Tandem Mass Spectrometry Data for Metabolite Identification and Structural Characterization. Anal Chem 80(16):6382-6389. doi:10.1021/ac800795f.
- Sturm M, Bertsch A, Gröpl C, Hildebrandt A, Hussong R, Lange E, Pfeifer N, Schulz-Trieglaff O, Zerck A, Reinert K, Kohlbacher O. OpenMS – an Open-Source Software Framework for Mass Spectrometry. BMC Bioinformatics. 2008;9:163. doi:10.1186/1471-2105-9-163.
- Libiseller G, Dvorzak M, Kleb U, Gander E, Eisenberg T, Madeo F, Neumann S, Trausinger G, Sinner F, Pieber T, Magnes C ((2015)). “IPO: a tool for automated optimization of XCMS parameters.” BMC Bioinformatics, 16, 118.
- Kai Dührkop, Huibin Shen, Marvin Meusel, Juho Rousu, and Sebastian Böcker. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc Natl Acad Sci U S A, 112(41):12580-12585, 2015. (cite this when using CSI:FingerID)
- Ruttkies C., Schymanski E.L. et al, MetFrag relaunched: incorporating strategies beyond in silico fragmentation. Journal of Cheminformatics, 2016, 8:3.
- Allen F, Greiner R, and Wishart D. Computational prediction of electron ionization mass spectra to assist in GC-MS compound identification. Submitted, 2016.
- Kerstin Scheubert, Franziska Hufsky, Daniel Petras, Mingxun Wang, Louis-Felix Nothias, Kai Dührkop, Nuno Bandeira, Pieter C Dorrestein, Sebastian Böcker. Significance estimation enabling large scale untargeted metabolomics annotations. Nature Communications, 2017. 8(1494)
Third party software copyright
nf-core/metaboigniter contains several scripts for performing various pre-processing steps.
The following software have not been written by us:
- MetFrag (GNU LGPL v2.1 or later). Files included:
bin/metfragbin/metfrag.jar
- jni-inchi (GNU LGPL v3). Files include:
bin/jni-inchi-0.8.jar
The rest of the scripts has been originally written by Payam Emami as part of PhenoMeNal (Phenome and Metabolome aNalysis) consortium.