nf-core/methylong
Extract methylation calls from long reads (ONT/ PacBio)
Introduction
nf-core/methylong is a bioinformatics pipeline that is tailored for long-read methylation calling. This pipeline requires a genome reference as input, and can take either modification-basecalled ONT reads, PacBio HiFi reads (modBam), raw sequencing Pod5 reads or raw Bam reads. The ONT workflow includes modcalling (optional), preprocessing (trim and repair) of reads, genome alignment and methylation calling. The PacBio HiFi workflow includes modcalling (optional), genome alignment and methylation calling. Methylation calls are extracted into BED/BEDGRAPH format, readily for direct downstream analysis. The downstream workflow includes SNV calling, phasing and DMR analysis.
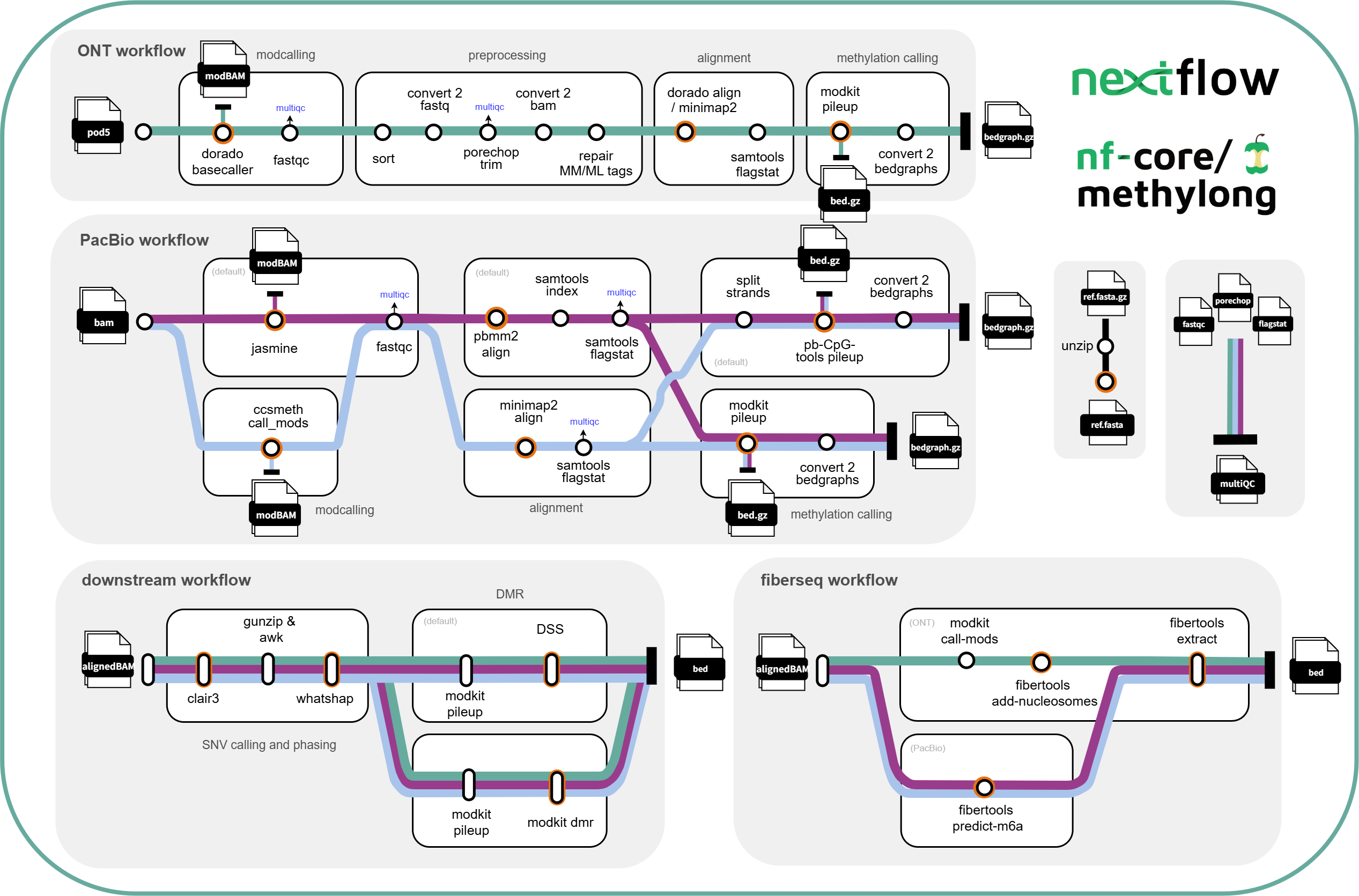
ONT workflow:
- modcalling (optional)
- basecall pod5 reads to modBam -
dorado basecaller sup --modified-bases 5mC_5hmC(default)
- basecall pod5 reads to modBam -
- trim and repair tags of input modBam
- trim and repair workflow:
- sort modBam -
samtools sort - convert modBam to fastq -
samtools fastq - trim barcode and adapters -
porechop - convert trimmed modfastq to modBam -
samtools import - repair MM/ML tags of trimmed modBam -
modkit repair
- sort modBam -
- trim and repair workflow:
- align to reference (plus sorting and indexing) -
dorado aligner( default) /minimap2- optional: remove previous alignment information before running
dorado alignerusingsamtools reset - include alignment summary -
samtools flagstat
- optional: remove previous alignment information before running
- create bedMethyl -
modkit pileup, 5x base coverage minimum. - create bedgraphs (optional)
PacBio workflow:
-
modcalling (optional)
- modcall bam reads to modBam -
jasmine(default) orccsmeth
- modcall bam reads to modBam -
-
align to reference -
pbmm2(default) orminimap2-
minimap workflow:
- convert modBam to fastq -
samtools convert - alignment -
minimap2 - sort and index -
samtools sort - alignment summary -
samtools flagstat
- convert modBam to fastq -
-
pbmm2 workflow:
- alignment and sorting -
pbmm2 - index -
samtools index - alignment summary -
samtools flagstat
- alignment and sorting -
-
-
create bedMethyl -
pb-CpG-tools(default) ormodkit pileup- notes about using
pb-CpG-toolspileup:- 5x base coverage minimum.
- 2 pile up methods available from
pb-CpG-tools:- default using
model - or
count(differences described here: https://github.com/PacificBiosciences/pb-CpG-tools)
- default using
pb-CpG-toolsby default merge mC signals on CpG into forward strand. To ‘force’ strand specific signal output, I followed the suggestion mentioned in this issue (PacificBiosciences/pb-CpG-tools#37) which uses HP tags to tag forward and reverse reads, so they were output separately.
- notes about using
-
create bedgraph (optional)
Downstream workflow:
- SNV calling -
clair3 - phasing -
whatshap phase - DMR analysis
- includes DMR haplotype level and population scale:
- tag reads by haplotype -
whatshap haplotype - create bedMethyl -
modkit pileup - DMR -
DSS(default) ormodkit dmr(default when--all-contextis set)- in
DSS, only regions with statistically significant CpG sites will be detected as DMRs.
- in
- tag reads by haplotype -
- includes DMR haplotype level and population scale:
Fiberseq workflow:
- ONT alignedBAM
- filtering m6A calls -
modkit call-mods - infer nucleosomes and MSPs -
ft add-nuleosomes - create bedMethyl -
ft extract
- filtering m6A calls -
- PacBio alignedBAM
- predict m6a and infer nucleosomes -
ft predict-m6a - create bedMethyl -
ft extract
- predict m6a and infer nucleosomes -
Usage
Currently no support of dorado and pb-CpG-tools through conda.
The pipeline can identify whether ONT reads are in pod5 or bam format, and automatically determine whether to perform basecalling.
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Required input:
- ONT or PacBio HiFi reads
- unaligned modification basecalled bam (modBam)
- if input modBam was aligned, remove previous alignment information using
--reset - raw ONT pod5
- raw bam
- reference genome
First, prepare a samplesheet with your input data that looks as follows:
group,sample,path,ref,methodtest,Col_0,ont_modbam.bam,Col_0.fasta,ont| Column | Content |
|---|---|
group |
Group of the sample |
sample |
Name of the sample |
path |
Path to sample file |
ref |
Path to assembly fasta/fa file |
method |
specify ont / pacbio |
Now, you can run the pipeline using:
nextflow run nf-core/methylong \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Folder stuctures of the outputs:
├── ont/sampleName│ ││ ├── fastqc│ ││ ├── basecall│ │ └── calls.bam│ ││ ├── fiberseq│ │ ├── m6acall.bam│ │ └── m6a.bed│ ││ ├── trim│ │ ├── trimmed.fastq.gz│ │ └── trimmed.log│ ││ ├── repair│ │ ├── repaired.bam│ │ └── repaired.log│ ││ ├── alignment│ │ ├── aligned.bam│ │ ├── aligned.bai│ │ └── aligned.flagstat│ ││ ├── snvcall│ │ ├── merge_output.vcf.gz│ │ ├── merge_output.vcf.gz.tbi│ │ └── SNV_PASS.vcf│ ││ ├── phase│ │ ├── phased.vcf.gz│ │ ├── haplotagged.bam│ │ └── haplotagged.readlist│ ││ ├── pileup│ │ ├── pileup.bed.gz│ │ └── pileup.log│ ││ ├── bedgraph│ │ └── <CG|CHG|CHH|A>.bedgraph.gz│ ││ ├── dmr_haplotype_level│ │ ├── phased_pileup│ │ │ ├── hp1.bed.gz│ │ │ ├── hp2.bed.gz│ │ │ └── combined.bed.gz│ │ └── dmr:modkit/dss│ │ ├── preprocessed_<1|2|etc>.bed│ │ ├── DSS_DMLtest.txt│ │ ├── DSS_callDML.txt│ │ ├── DSS_callDMR.txt│ │ └── DSS.log│ │ ├── group1_group2_modkit.bed.gz│ │ └── group1_group2_modkit_dmr.log│ ││ └── dmr_population_scale│ ├── group_pileup│ │ ├── group1.bed.gz│ │ └── group2.bed.gz│ └── group1_group2:dmr:modkit/dss│ ├── population_scale_DMLtest.txt│ ├── population_scale_callDML.txt│ ├── population_scale_callDMR.txt│ └── population_scale.log│ ├── group1_group2_modkit.bed.gz│ └── group1_group2_modkit_dmr.log││├── pacbio/sampleName│ ││ ├── fastqc│ ││ ├── modcall│ │ └── modbam.bam│ ││ ├── fiberseq│ │ ├── m6a_predicted.bam│ │ └── m6a.bed│ ││ ├── alignment│ │ ├── aligned.bam│ │ ├── aligned.bai/csi│ │ └── aligned.flagstat│ ││ ├── pileup: modkit/pb_cpg_tools│ │ ├── pileup.bed.gz│ │ ├── pileup.log│ │ └── pileup.bw (only pb_cpg_tools)│ ││ ├── snvcall│ │ ├── merge_output.vcf.gz│ │ └── SNV_PASS.vcf│ ││ ├── phase│ │ ├── phased.vcf.gz│ │ ├── haplotagged.bam│ │ └── haplotagged.readlist│ ││ ├── bedgraph│ │ └── bedgraphs│ ││ ├── dmr_haplotype_level│ │ ├── phased_pileup│ │ │ ├── hp1.bed.gz│ │ │ ├── hp2.bed.gz│ │ │ └── combined.bed.gz│ │ └── dmr:modkit/dss│ │ ├── preprocessed_<1|2|etc>.bed│ │ ├── DSS_DMLtest.txt│ │ ├── DSS_callDML.txt│ │ ├── DSS_callDMR.txt│ │ └── DSS.log│ │ ├── group1_group2_modkit.bed.gz│ │ └── group1_group2_modkit_dmr.log│ ││ └── dmr_population_scale│ ├── group_pileup│ │ ├── group1.bed.gz│ │ └── group2.bed.gz│ └── group1_group2:dmr:modkit/dss│ ├── population_scale_DMLtest.txt│ ├── population_scale_callDML.txt│ ├── population_scale_callDMR.txt│ └── population_scale.log│ ├── group1_group2_modkit.bed.gz│ └── group1_group2_modkit_dmr.log│└── multiqc │ ├── fastqc └── flagstatbedgraph outputs all have min. 5x base coverage.
Credits
nf-core/methylong was originally written by Jin Yan Khoo, from the Faculty of Biology of the Ludwig-Maximilians University (LMU) in Munich, Germany, funded by in part via TRR356 (DFG Grant Number, 491090170, Project A05 to Niklas Schandry). Further significant contributions were made by YiJin Xiong, from Central South University (CSU) in Changsha, China.
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #methylong channel (you can join with this invite).
Citations
If you use nf-core/methylong for your analysis, please cite it using the following doi: 10.5281/zenodo.15366448
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.