nf-core/nanoseq
Nanopore demultiplexing, QC and alignment pipeline
Introduction
This document describes the output produced by the nf-core/nanoseq pipeline. Most of the plots shown here are taken from the MultiQC report, which summarises the pipeline results.
Pipeline overview
The nf-core/nanoseq pipeline is built using Nextflow. There are many different potential outputs for the pipeline depending on what file inputs and parameters you use. Please see main README.md for a condensed overview of the steps in the pipeline and the bioinformatics tools used at each step.
See Oxford NanoPore website for more information regarding the sequencing technology, protocol, and for an extensive list of additional resources.
The directories listed below are created in the output directory after the pipeline has finished. All paths are relative to the top-level results directory.
Demultiplexing
Output files
qcat/fastq/<barcode*>.fastq.gz: FASTQ output files for each barcode.qcat/fastq/none.fastq.gz: FASTQ file with reads were unassigned to any given barcode.
Documentation: qcat
Description:
Nanoseq is designed to deal with various input data types. Pre-basecalled FASTQ file can be demuliplexed using qcat. Please see usage.md for more details about the format of the input samplesheet. All potential outputs are shown here however some may not be produced depending on your inputs and the steps you have chosen to run.
Removal of DNA contaminants
Output files
nanolyse/<SAMPLE>.fastq.gz: FASTQ file after the removal of reads that map to DNA contaminants.nanolyse/<SAMPLE>.nanolyse.log: NanoLyse log file.
Documentation: NanoLyse
Description:
If you would like to run NanoLyse on the raw FASTQ files you can provide --run_nanolyse when running the pipeline. By default, the pipeline will filter lambda phage reads. However, you can provide your own FASTA file of “contaminants” with --nanolyse_fasta. The filtered FASTQ files will contain raw reads without the specified reference sequences (default: lambda phage sequences).
Read QC
Output files
nanoplot/fastq/<SAMPLE>/: directory with various*.htmlfiles containing QC metrics and plots.fastqc/<SAMPLE>_fastqc.html: FastQC*.htmlfile for each sample.fastqc/<SAMPLE>_fastqc.zip: FastQC*.zipfile for each sample.
Documentation: NanoPlot, FastQC
Description: NanoPlot can be used to produce general quality metrics from the per barcode FASTQ files generated by a basecaller e.g. quality score distribution, read lengths, and other general stats.
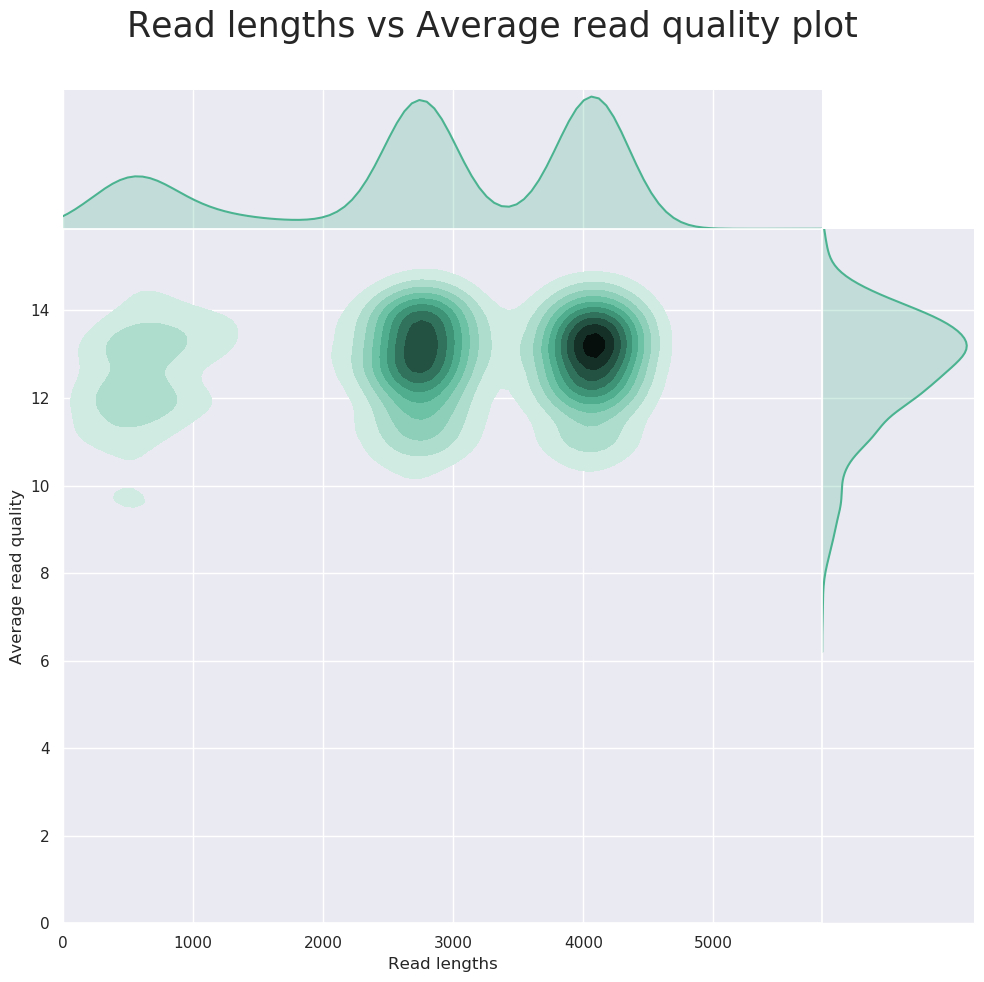
FastQC can give general quality metrics about your reads. It can provide information about the quality score distribution across your reads, and the per-base sequence content (%A/C/G/T). You can also generate information about adapter contamination and other over-represented sequences.
Alignment
Output files
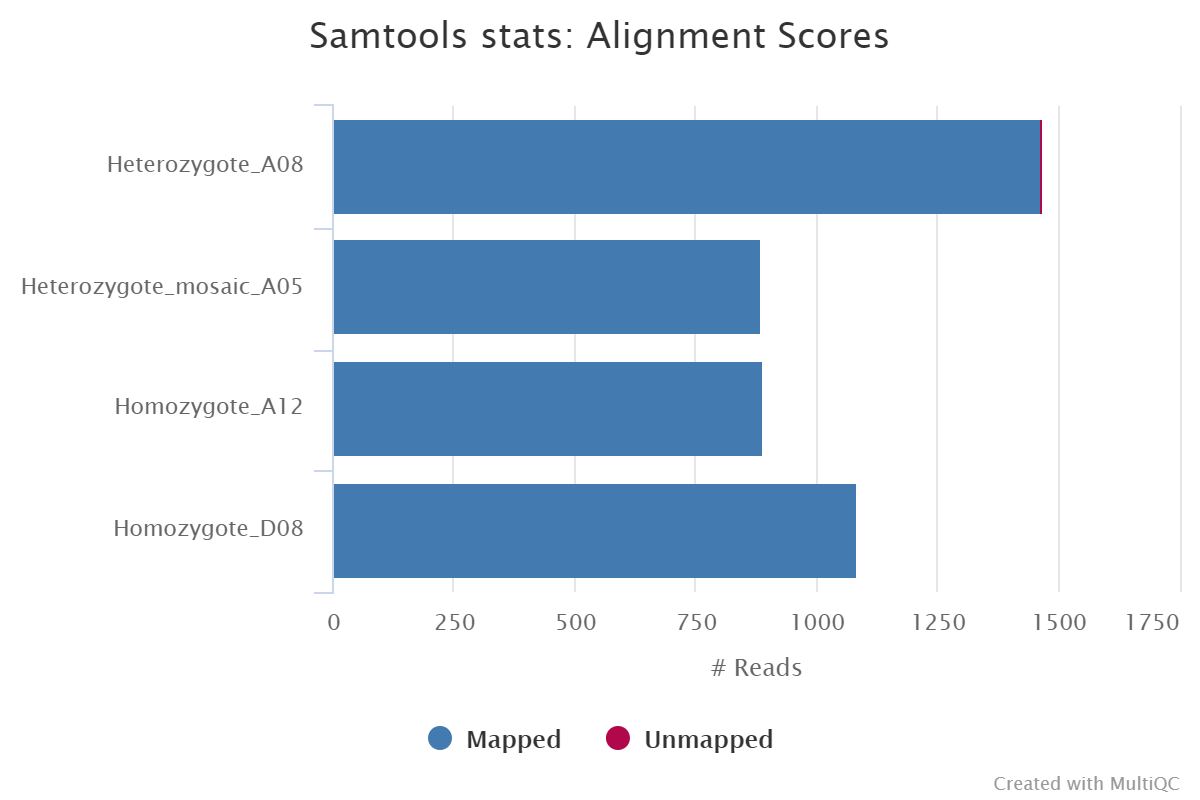
<ALIGNER>/<SAMPLE>.sorted.bam: per sample coordinate sorted BAM file.<ALIGNER>/<SAMPLE>.sorted.bam.bai: per sample coordinate sorted BAM index file.<ALIGNER>/samtools_stats/: directory with per sample*.flagstat,*.idxstatsand*.statsfiles generated by Samtools.
Documentation: GraphMap2, MiniMap2, Samtools
Description:
Reads are mapped to a user-defined genome or transcriptome using either GraphMap2 or Minimap2. The resulting BAM file is sorted and indexed using Samtools. If the same reference is specified multiple times in the input sample sheet then the aligner index will only be built once for re-use across all samples. You can skip the alignment and downstream processes by providing the --skip_alignment parameter.
The initial SAM alignment file created by GraphMap2 or Minimap2 are not saved by default to be more storage space efficient. You can override this behaviour by using the --save_align_intermeds parameter.
Coverage tracks
Output files
<ALIGNER>/bigwig/: directory with per sample*.bigWigand*.bedGraphfile.<ALIGNER>/bigbed/: directory with per sample*.bigBedand*bed12file.
Documentation:
BEDTools, bedGraphToBigWig, bedToBigBed
Description: The bigWig format is an indexed binary format useful for displaying dense, continuous data in Genome Browsers such as the UCSC and IGV. This mitigates the need to load the much larger BAM file for data visualisation purposes which will be slower and result in memory issues. The bigWig format is also supported by various bioinformatics software for downstream processing such as meta-profile plotting.
bigBed are more useful for displaying distribution of reads across exon intervals as is typically observed for RNA-seq data. Therefore, these files will only be generated if --protocol directRNA or --protocol cDNA are defined.
The creation of these bigwig and bigbed files can be bypassed by setting the --skip_bigwig and --skip_bigbed parameters, respectively.
Variant calling
Output files
Short variant callers
-
Medaka:
variant_calling/medaka_variant/<SAMPLE>.vcf.gz: zipped VCF file with small variants.variant_calling/medaka_variant/<SAMPLE>.vcf.gz: index for zipped VCF file with small variants.
-
DeepVariant:
variant_calling/deepvariant/<SAMPLE>.vcf.gz: zipped VCF file with small variants.variant_calling/deepvariant/<SAMPLE>.vcf.gz.tbi: index for zipped VCF file with small variants.
-
PEPPER-Margin-DeepVariant:
variant_calling/margin_pepper_deepvariant/<SAMPLE>.vcf.gz: zipped VCF file with small variants.variant_calling/margin_pepper_deepvariant/<SAMPLE>.vcf.gz.tbi: index for zipped VCF file with small variants.
Structural variant callers
-
Sniffles
variant_calling/cutesv/<SAMPLE>.vcf.gz: zipped VCF file with smtructural variants.variant_calling/cutesv/<SAMPLE>.vcf.gz.tbi: index for zipped VCF file with structural variants.
-
cuteSV
variant_calling/sniffles/<SAMPLE>.vcf.gz: zipped VCF file with smtructural variants.variant_calling/sniffles/<SAMPLE>.vcf.gz.tbi: index for zipped VCF file with structural variants.
Documentation: Medaka, DeepVariant, PEPPER-Margin-DeepVariant, Sniffles, cuteSV
Description:
If the --protocol DNA and the --call_variants parameters are defined then both small and structural variant variant calls can be generated.
Short variants can be called using medaka, deepvariant or pepper_margin_deepvariant. The short variant caller is specified using the --variant_caller parameter.
Structural variants can be called using either cuteSV or sniffles. The structural variant caller is specified using the --structural_variant_caller parameter.
The short variant and/or structural variant calling steps is skipped if using the --skip_vc and --skip_sniffles flags.
Transcript Reconstruction and Quantification
Output files
If bambu is used:
bambu/extended_annotations.gtf: a GTF file that contains both annotated and novel transcripts.counts_gene.txt: a TXT file containing gene expression estimates.counts_transcript.txt: a TXT file containing transcript expression estimates.
If StringTie2 is used:
stringtie2/*.bam: per sample coordinate sorted alignment file.*.stringtie.gtf: per sample annotations for novel transcripts obtained in StringTie2.stringtie.merged.gtf: extended annotation that combines provided GTF with GTF file from each sample via StringTie2 Merge.counts_gene.txt: gene expression estimates calculated by featureCounts.counts_gene.txt.summary: FeatureCounts gene level log file.counts_transcript.txt: transcript expression estimates calculated by featureCounts.counts_transcript.txt.summary: FeatureCounts transcript level log file.
Documentation: bambu, StringTie2, featureCounts
Description:
After genomic alignment, novel transcripts can be reconstructed using tools such as bambu and StringTie2. Quantification can then be performed on a more complete annotation based on the transcripts detected within a given set of samples. bambu performs both the reconstruction and quantification steps. An an alternative approach, we also provides an option to run StringTie2 to identify novel transcripts. However, when multiple samples are provided, quantification for multiple samples are not implemented explicitly in the software. Hence a second step is required to merge novel transcripts across multiple samples followed by quantification for both gene and transcripts using featureCounts. You can skip transcript reconstruction and quantification by providing the --skip_quantification parameter.
Differential expression analysis
Output files
<QUANTIFICATION_METHOD>/deseq2/deseq2.results.txt: a TXT file that contains differential gene expression.<QUANTIFICATION_METHOD>/dexseq/dexseq.results.txt: a TXT file that contains differential transcript expression.
Description:
If multiple conditions and multiple replicates are available then the pipeline is able to run differential analysis on gene and transcripts with DESeq2 and DEXSeq, respectively. These steps won’t be run if you provide the --skip_quantification or --skip_differential_analysis parameters or if all of the samples in the samplesheet don’t have the same FASTA and GTF reference files.
RNA modification analysis
Output files
rna_modifications/xpore/diffmod/diffmod_outputs/diffmod.table: a table file that contains differentially modified sites.rna_modifications/m6anet/inference/<sample_name>/data.result.csv.gz: a CSV file that contains m6A sites.
Description:
If multiple conditions are available then the pipeline is able to run differential modification analysis with xPore. These steps won’t be run if you provide the --skip_modification_analysis or --skip_xpore or --skip_m6anet parameters.
RNA fusion analysis
Output files
jaffal/jaffa_results.csv: a CSV file that contains RNA fusion results.jaffal/jaffa_results.fasta: a FASTA file that contains the sequence of the RNA fusions.
Documentation: jaffal
Description:
This step won’t be run if you provide the --skip_fusion_analysis parameter.
MultiQC
Output files
multiqc/multiqc_report.html: a standalone HTML file that can be viewed in your web browser.multiqc_data/: a directory containing parsed statistics from the different tools used in the pipeline.multiqc_plots/: a directory containing static images from the report in various formats.
Documentation: MultiQC
Description: MultiQC is a visualisation tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available within the report data directory.
Results generated by MultiQC for this pipeline collate QC from FastQC, samtools flagstat, samtools idxstats and samtools stats.
The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability. For more information about how to use MultiQC reports, see http://multiqc.info.
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*file will only be present if the--email/--email_on_failparameter’s are used when running the pipeline. - Reformatted samplesheet file used as the pipeline input:
samplesheet.valid.csv.
- Reports generated by Nextflow:
Documentation: Nextflow
Description: Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to trouble-shoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.