nf-core/oncoanalyser
A comprehensive cancer DNA/RNA analysis and reporting pipeline
Introduction
nf-core/oncoanalyser is a Nextflow pipeline for the comprehensive analysis of cancer DNA and RNA sequencing data using the WiGiTS toolkit from the Hartwig Medical Foundation. The pipeline supports a wide range of experimental setups:
- FASTQ, BAM, and / or CRAM input files
- WGS (whole genome sequencing), WTS (whole transcriptome sequencing), and targeted / panel sequencing1
- Paired tumor / normal and tumor-only samples, and support for donor samples for further normal subtraction
- Purity estimate for longitudinal samples using genomic features of the primary sample from the same patient2
- UMI (unique molecular identifier) processing supported for DNA sequencing data
- Most GRCh37 and GRCh38 reference genome builds
1 built-in support for the TSO500
panel with other
panels and exomes requiring creation of custom panel reference
data
2 for example a primary WGS tissue biospy and longitudinal low-pass WGS ccfDNA sample taken from the
same patient
Pipeline overview
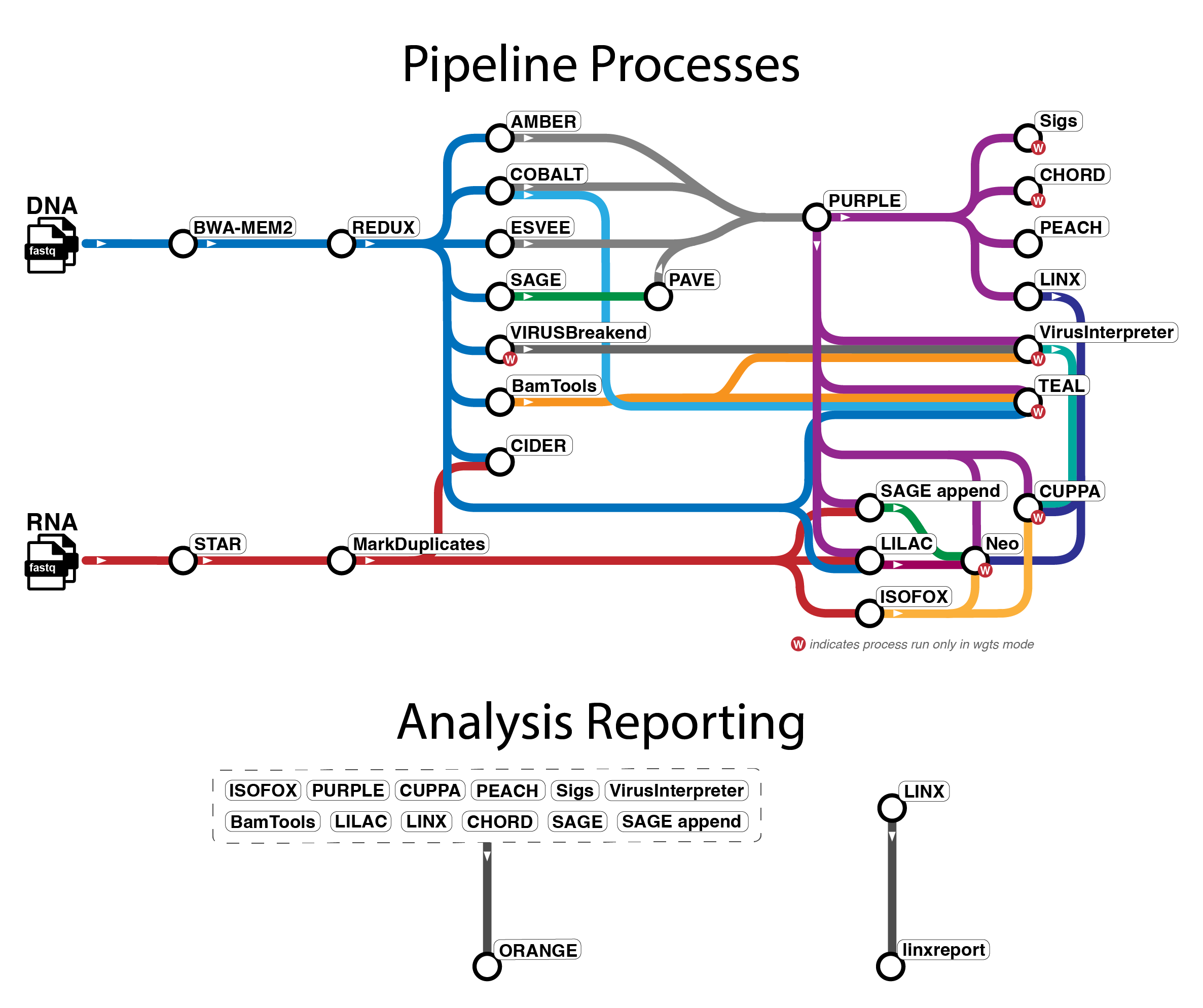
The pipeline mainly uses tools from WiGiTS, as well as some other external
tools. There are several workflows available in oncoanalyser and
the tool information below primarily relates to the wgts and targeted analysis modes.
Due to the limitations of panel data, certain tools (indicated with * below) do not run in targeted mode.
- Read alignment: BWA-MEM2 (DNA), STAR (RNA)
- Read post-processing: REDUX (DNA), Picard MarkDuplicates (RNA)
- SNV, MNV, INDEL calling: SAGE, PAVE
- SV calling: ESVEE
- CNV calling: AMBER, COBALT, PURPLE
- SV and driver event interpretation: LINX
- RNA transcript analysis: ISOFOX
- Oncoviral detection: VIRUSbreakend*, VirusInterpreter*
- Telomere characterisation: TEAL*
- Immune analysis: LILAC, CIDER, NEO*
- Mutational signature fitting: SIGS*
- HRD prediction: CHORD*
- Tissue of origin prediction: CUPPA*
- Pharmacogenomics: PEACH
- Summary report: ORANGE, linxreport
For the purity_estimate mode, several of the above tools are run with adjusted configuration in addition to the following.
- Tumor fraction estimation: WISP
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Create a samplesheet with your inputs (WGS/WTS BAMs in this example):
group_id,subject_id,sample_id,sample_type,sequence_type,filetype,filepath
PATIENT1_WGTS,PATIENT1,PATIENT1-N,normal,dna,bam,/path/to/PATIENT1-N.dna.bam
PATIENT1_WGTS,PATIENT1,PATIENT1-T,tumor,dna,bam,/path/to/PATIENT1-T.dna.bam
PATIENT1_WGTS,PATIENT1,PATIENT1-T-RNA,tumor,rna,bam,/path/to/PATIENT1-T.rna.bamLaunch oncoanalyser:
nextflow run nf-core/oncoanalyser \
-profile <docker/singularity/.../institute> \
-revision 2.3.0 \
--mode <wgts/targeted> \
--genome <GRCh37_hmf/GRCh38_hmf> \
--input samplesheet.csv \
--outdir output/Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Version information
Extended support
As oncoanalyser is used in clinical settings and subject to accreditation standards in some instances, there is a need
for long-term stability and reliability for feature releases in order to meet operational requirements. This is
accomplished through long-term support of several nominated feature releases, which all receive bug fixes and security
fixes during the period of extended support.
Each release that is given extended support is allocated a separate long-lived git branch with the ‘stable’ prefix, e.g.
stable/1.2.x, stable/1.5.x. Feature development otherwise occurs on the dev branch with stable releases pushed to
master.
Versions nominated to have current long-term support:
- TBD
Known issues
Please refer to this page for details regarding any known issues.
Credits
The oncoanalyser pipeline was written and is maintained by Stephen Watts (@scwatts) from
the Genomics Platform
Group at the University
of Melbourne Centre for Cancer Research.
We thank the following organisations and people for their extensive assistance in the development of this pipeline, listed in alphabetical order:
- Hartwig Medical Foundation Australia
- Oliver Hofmann
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #oncoanalyser
channel (you can join with this invite).
Citations
You can cite the oncoanalyser Zenodo record for a specific version using the following DOI:
10.5281/zenodo.15189386
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md
file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.