nf-core/pacvar
Longread PacBio sequencing processing for WGS and PureTarget
Introduction
nf-core/pacvar is a bioinformatics pipeline that processes long-read PacBio data. Specifically, the pipeline provides two workflows: one for processing whole-genome sequencing data, and another for processing reads from the PureTarget expansion panel offered by PacBio. This second workflow characterizes tandem repeats. Because the pipeline is designed for PacBio reads, it uses PacBio’s officially released tools.
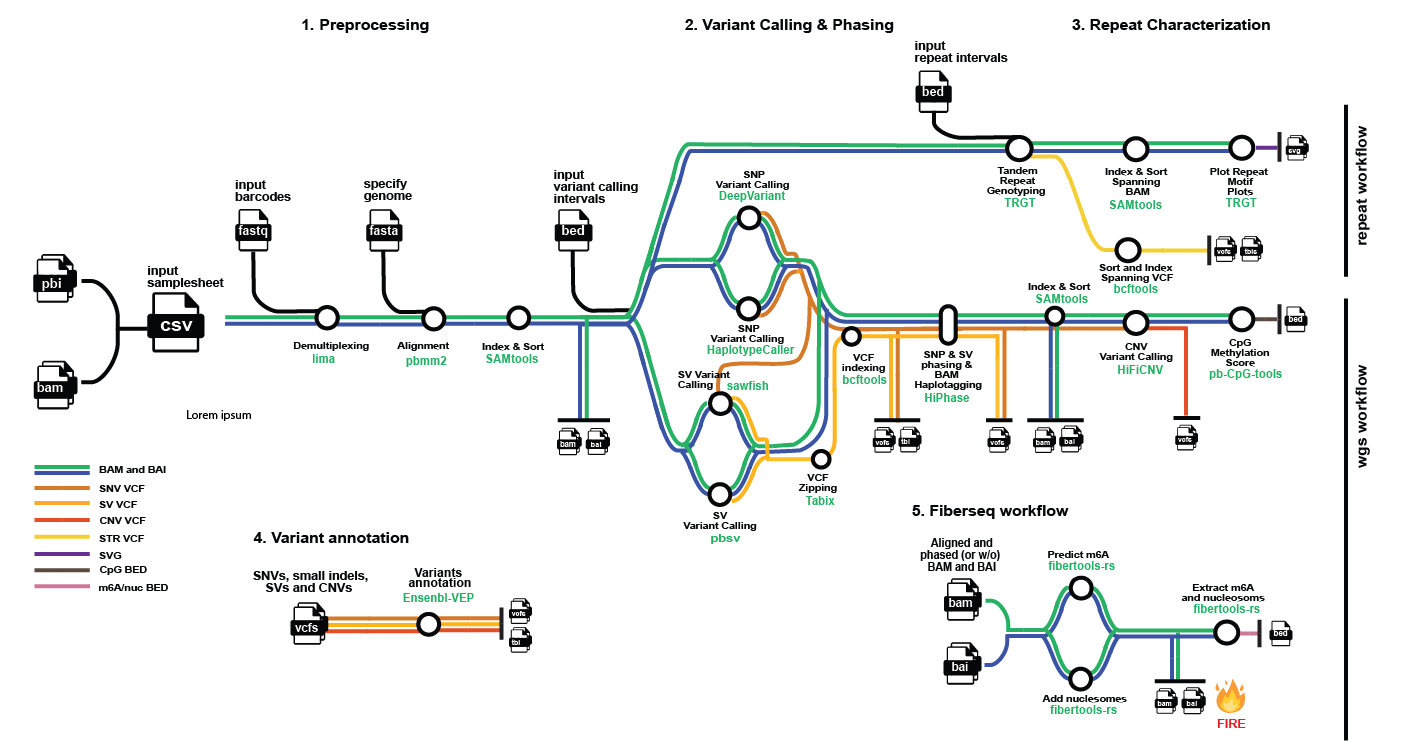
Preprocessing Overview
WGS Workflow Overview
- Choice of SNVs and small indels calling routes:
a.
DeepVariant(default) b.HaplotypeCaller - Choice of SV calling routes:
a.
sawfish(default) b.pbsv - Index
pbsv’s VCF files (bcftools) - Phase SNVs, SVs and BAM files (
hiphase) - CNV calling (
HiFiCNV) - Extracts per-CpG methylation scores (
pb-CpG-tools::aligned_bam_to_cpg_scores) - SNV, small indel, SV, and CNV annotation with Ensembl VEP
Because sawfish consolidates both SV and CNV-related events, users may optionally disable the HiFiCNV step using --skip_hificnv true when sawfish is selected as the SV caller to avoid redundant CNV analyses.
The Ensembl VEP integration in this pipeline does not bundle plugins or custom files. Also, the current VEP cache (115) does not support the CHM13 homo sapiens genome. If using CHM13 for the wgs workflow, disable VEP using --skip_ensemblvep true. When using the default S3 VEP cache, avoid adding --merged or --refseq to custom VEP arguments because the cache does not include the additional files required by these options.
Fiber-seq Workflow Overview
Set --skip_fiberseq false to extend the WGS workflow with Fiber-seq processing.
- Predict m6A calls (
fibertools-rs::predict-m6a) - Add nucleosome/MSP BAM auxiliary tags (
fibertools-rs::addnucleosome) - Extracts nucleosome positions (
fibertools-rs::extract)
For Fiber-seq pre-processing, set --skip_fiberseq false to run fibertools-rs m6A/nucleosome processing as part of the WGS workflow. Use --skip_m6A_predict false when the BAM contains PacBio kinetic tags (ip/pw or fi/fp/ri/rp) and needs m6A prediction; use --skip_m6A_predict true when the BAM already contains A+a m6A tags and only nucleosome/MSP annotation is needed.
When using the Fiber-seq workflow, it is highly recommended to run SNV calling and HiPhase so Fiber-seq processing uses a phased BAM when available. Haplotype-phased BAMs preserve long-read haplotype context, helping downstream FIRE analyses take full advantage of long-read sequencing when inferring regulatory elements.
Tandem Repeat Workflow Overview
- Genotype tandem repeats - produce spanning bams and vcf (
TRGT) - Index and Sort tandem tepeat spanning bam (
SAMtools) - Plot repeat motif plots (
TRGT) - Sort spanning VCF (
bcftools)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,bam,pbiCONTROL,AEG588A1_S1_L002_R1_001.bam,AEG588A1_S1_L002_R1_001.pbiNote that the .pbi file is not required. If you choose not to include it, your input file might look like this:
sample,bam,pbiCONTROL,AEG588A1_S1_L002_R1_001.bamEach row represents an unaligned bam file and their associated index (optional).
For the repeat workflow, you can optionally provide a fail BAM in the samplesheet. When a fail BAM is provided, the repeat workflow merges the mapped HiFi and fail BAMs after mapping, before repeat-specific analysis:
sample,bam,pbi,failCONTROL,AEG588A1_S1_L002_R1_001.bam,AEG588A1_S1_L002_R1_001.pbi,AEG588A1_S1_fail.bamNow, you can run the pipeline. Below is an example
nextflow run nf-core/pacvar \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --workflow <wgs/repeat> \ --barcodes barcodes.bed \ --intervals intervals.bed \ --genome <GENOME NAME (e.g. GATK.GRCh38)> \ --outdir <OUTDIR>Optional paramaters include: --skip_demultiplexing, --skip_snp, --skip_sv, --skip_phase, --skip_hificnv, --skip_cpg, --skip_fiberseq, --skip_m6A_predict, and --skip_ensemblvep. The variant callers can be specified using --snv_caller <deepvariant/haplotypecaller> and --sv_caller <sawfish/pbsv>.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/pacvar was originally written by Tanya Sarkin Jain. Contributions by Chao-Jen Wong and Stijn Van de Sompele were added starting with version 1.1.0dev and continuing in later releases.
We thank the following people for their extensive assistance in the development of this pipeline:
- Evangelos Karatzas for his meticulous review involving 73 conversation threads.
- Tania Jain for providing comments and guidance throughout the v1.1.0 development phase.
- The Seqera and nf-core communities: Many members have contributed by reviewing modules and pull requests. This pipeline is a community effort.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #pacvar channel (you can join with this invite).
Citations
If you use nf-core/pacvar for your analysis, please cite it using the following doi: 10.5281/zenodo.14813048 and also publication:
Tanya Jain, Claire Clelland, nf-core/pacvar: a pipeline for analyzing longread PacBio whole genome and repeat expansion sequencing data, Bioinformatics, 2025;, btaf116, https://doi.org/10.1093/bioinformatics/btaf116
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.