nf-core/rnafusion
RNA-seq analysis pipeline for detection of gene-fusions
1.0). The latest
stable release is
3.0.1b
.

![]()

![]()

![]()
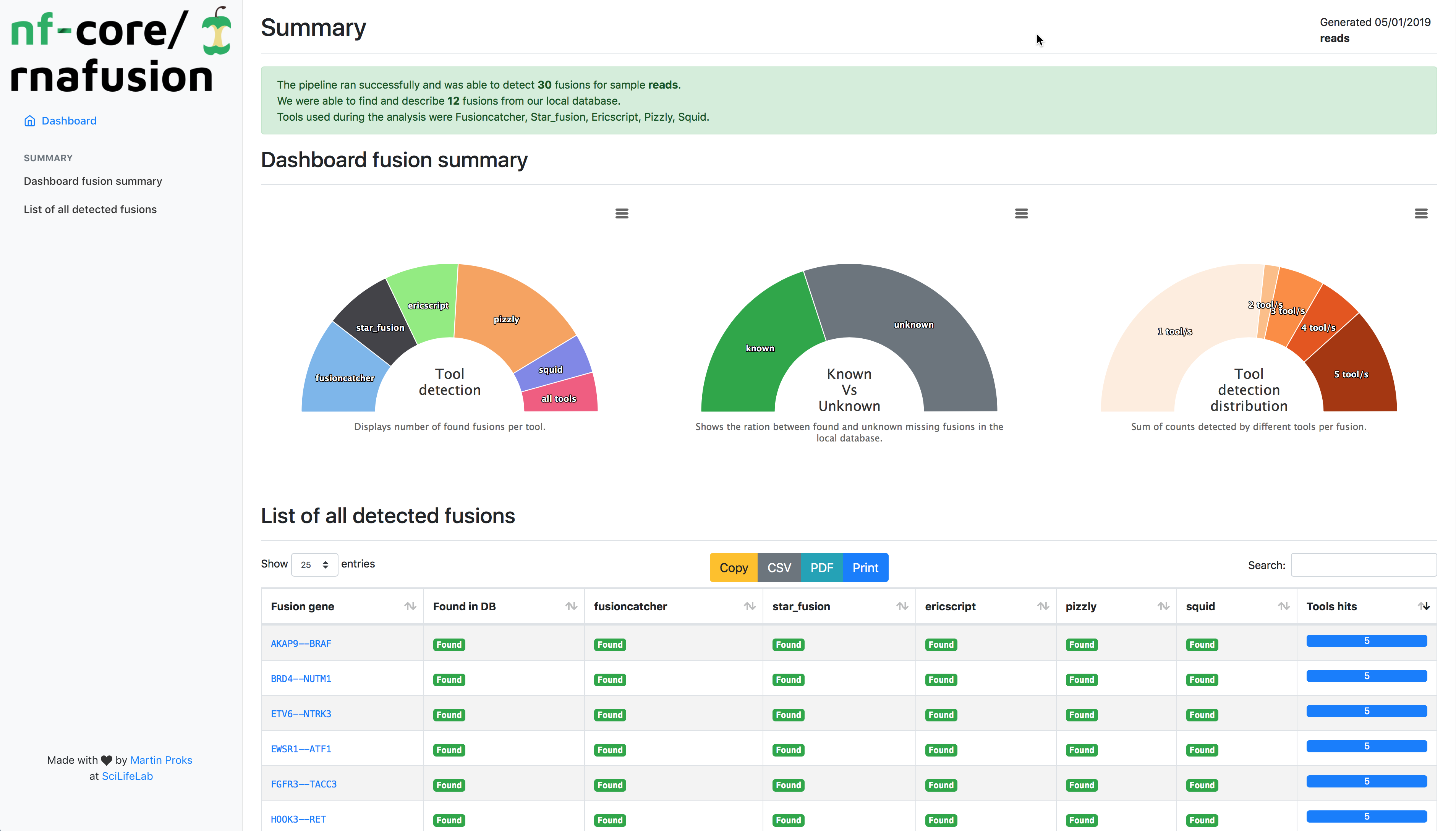
nfcore/rnafusion uses RNA-seq data to detect fusions genes.
The workflow processes RNA-sequencing data from FastQ files. It runs quality control on the raw data (FastQC), detects fusion genes (STAR-Fusion, Fusioncatcher, Ericscript, Pizzly, Squid), gathers information (FusionGDB), visualizes the fusions (FusionInspector), performs quality-control on the results (MultiQC) and finally generates custom summary report.
The pipeline works with both single-end and paired-end data, though not all fusion detection tools work with single-end data (Ericscript, Pizzly, Squid and FusionInspector).
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker / singularity containers making installation trivial and results highly reproducible.
| Tool | Single-end reads | CPU (recommended) | RAM (recommended) |
|---|---|---|---|
| Star-Fusion | Yes | >=16 cores | ~30GB |
| Fusioncatcher | Yes | >=16 cores | ~60GB |
| Ericscript | No | >=16 cores | ~30GB |
| Pizzly | No | >=16 cores | ~30GB |
| Squid | No | >=16 cores | ~30GB |
| FusionInspector | No | >=16 cores | ~30GB |
TL;DR: Make sure to download all required references for each tool. More details can be found in section tools.
nextflow run nf-core/rnafusion --reads '*_R{1,2}.fastq.gz' --genome GRCh38 -profile docker --star_fusion --fusioncatcher --ericscript --pizzly --squid --fusion_inspectorFor available parameters or help run:
nextflow run nf-core/rnafusion --helpDocumentation
The nf-core/rnafusion pipeline comes with documentation about the pipeline, found in the docs/ directory:
- Installation
- Pipeline configuration
- Running the pipeline
- Output and how to interpret the results
- Troubleshooting
Use predefined configuration for desired Institution cluster provided at nfcore/config repository.