nf-core/rnasplice
rnasplice is a bioinformatics pipeline for RNA-seq alternative splicing analysis
Introduction
nf-core/rnasplice is a bioinformatics pipeline for alternative splicing analysis of RNA sequencing data obtained from organisms with a reference genome and annotation.
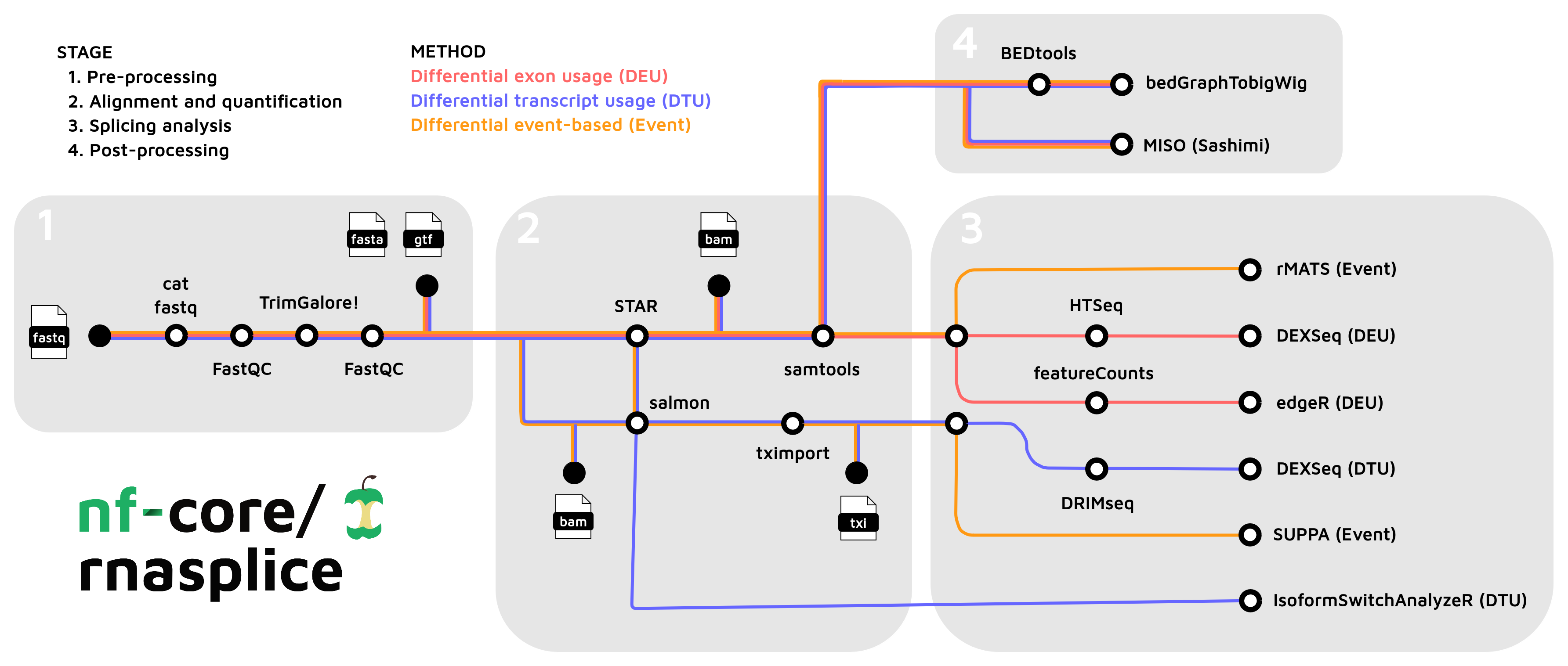
- Merge re-sequenced FastQ files (
cat) - Read QC (
FastQC) - Adapter and quality trimming (
TrimGalore) - Alignment with
STAR - Choice of quantification depending on analysis type:
- Sort and index alignments (
SAMtools) - Create bigWig coverage files (
BEDTools,bedGraphToBigWig) - Pseudo-alignment and quantification (
Salmon; optional) - Summarize QC (
MultiQC) - Differential Exon Usage (DEU):
HTSeq->DEXSeqfeatureCounts->edgeR- Quantification with
featureCountsorHTSeq - Differential exon usage with
DEXSeqoredgeR
- Differential Transcript Usage (DTU):
Salmon->DRIMSeq->DEXSeq- Filtering with
DRIMSeq - Differential transcript usage with
DEXSeq Salmon->IsoformSwitchAnalyzeR- Isoform switch analysis with
IsoformSwitchAnalyzeR
- Event-based splicing analysis:
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,fastq_1,fastq_2,strandedness,conditionCONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,forward,CONTROLCONTROL_REP1,AEG588A1_S1_L003_R1_001.fastq.gz,AEG588A1_S1_L003_R2_001.fastq.gz,forward,CONTROLCONTROL_REP1,AEG588A1_S1_L004_R1_001.fastq.gz,AEG588A1_S1_L004_R2_001.fastq.gz,forward,CONTROLEach row represents a fastq file (single-end) or a pair of fastq files (paired end). Rows with the same sample identifier are considered technical replicates and merged automatically. The strandedness refers to the library preparation and should be specified by the user.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those
provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
Now, you can run the pipeline using:
nextflow run nf-core/rnasplice \ --input samplesheet.csv \ --contrasts contrastsheet.csv \ --genome GRCh37 \ --outdir <OUTDIR> \ -profile <docker/singularity/.../institute>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Online videos
You can find numerous talks on the nf-core events page from various topics including writing pipelines/modules in Nextflow DSL2, using nf-core tooling, running nf-core pipelines as well as more generic content like contributing to Github. Please check them out!
Credits
nf-core/rnasplice was originally written by the bioinformatics team from Zifo RnD Solutions:
- Benjamin Southgate
- James Ashmore
- Valentino Ruggieri
- Claire Prince
- Keerthana Bhaskaran
- Asma Ali
- Lathika Madhan Mohan
We thank Harshil Patel (@drpatelh), Seqera Labs (seqeralabs) and Jesse Angelis (@jesseangelis) for their assistance in the development of this pipeline.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #rnasplice channel (you can join with this invite).
Citations
If you use nf-core/rnasplice for your analysis, please cite it using the following doi: 10.5281/zenodo.8424632
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.