nf-core/seqinspector
Dedicated QC-only pipeline for sequencing data. The pipeline will run a (potentially large) set of QC tools and can output global and group specific Multiqc reports. The pipeline is targeting core facilities or research groups with larger sequencing throughput.
Introduction
nf-core/seqinspector is a bioinformatics pipeline that processes raw sequence data (FASTQ) to provide comprehensive quality control. It can perform subsampling, quality assessment, duplication level analysis, and complexity evaluation on a per-sample basis, while also detecting adapter content, technical artifacts, and common biological contaminants. The pipeline generates detailed MultiQC reports with flexible output options, ranging from individual sample reports to project-wide summaries, making it particularly useful for sequencing core facilities and research groups with access to sequencing instruments. If provided, nf-core/seqinspector can also parse statistics from an Illumina run folder directory into the final MultiQC reports.
Compatibility between tools and data type
| Tool Type | Tool Name | Tool Description | Compatibility with Data | Dependencies | Default tool |
|---|---|---|---|---|---|
Subsampling |
Seqtk |
Global subsampling of reads. Only performs subsampling if --sample_size parameter is given. |
[RNA, DNA] | [N/A] | no |
Lint FASTQs |
fq |
fq filters, generates, subsamples, and validates FASTQ files. | [RNA, DNA, synthetic] | [N/A] | yes |
Trimming |
Chelae |
Adapter and quality trimming of short-read FASTQ data. | [RNA, DNA, synthetic] | [N/A] | no |
Trimming |
Fastp |
Trimming of reads. Only performs trimming if --tools parameter is given. |
[RNA, DNA, synthetic] | [N/A] | no |
Deduplication assessment |
BBMap Clumpify |
Deduplicate and compress FASTQ files. Only performs clumpify if --tools parameter is given. |
[RNA, DNA] | [N/A] | no |
Indexing, Mapping |
BWA-MEM2 |
Align reads to reference | [RNA, DNA] | [N/A] | yes |
Indexing |
SAMtools |
Index aligned BAM files, create FASTA index | [DNA] | [N/A] | yes |
QC |
checkQC |
Read QC | [RNA, DNA] | Illumina rundir | no |
QC |
FastQC |
Read QC | [RNA, DNA] | [N/A] | yes |
QC |
FASTQE |
Read QC | [RNA, DNA] | [N/A] | no |
QC |
FastqScreen |
Basic contamination detection | [RNA, DNA] | [N/A] | yes |
QC |
SeqFu Stats |
Sequence statistics | [RNA, DNA] | [N/A] | yes |
QC |
Seqkit Stats |
Simple statistics of FASTA/Q files | [RNA, DNA] | [N/A] | no |
QC |
Sequali |
Read QC for long and short reads. | [RNA, DNA] | [N/A] | yes |
Taxonomic Classification |
Kraken2 |
Performs taxonomic classification and/or profiling | [RNA, DNA] | [N/A] | no |
QC |
Picard CollectMultipleMetrics |
Collect multiple QC metrics | [RNA, DNA] | [BWA-MEM2, SAMtools, --genome] |
yes |
QC |
Picard CollectHsMetrics |
Collect alignment QC metrics of hybrid-selection data. | [RNA, DNA] | [BWA-MEM2, SAMtools, --fasta, --bait_intervals, --target_intervals (--ref_dict)] |
no |
QC |
Riker |
Collect BAM-level QC metrics (alignment, base distribution, insert size, GC bias, WGS, error metrics). | [DNA] | [BWA-MEM2, SAMtools, --fasta] |
no |
Reporting |
MultiQC |
Present QC for raw reads | [RNA, DNA, synthetic] | [N/A] | yes |
Reporting |
Krona |
Plotting Kraken2 results | [RNA, DNA, synthetic] | [kraken2] | no |
Workflow diagram
The subway map below is a graphical representation of the pipeline workflow, generated with nf-metro.
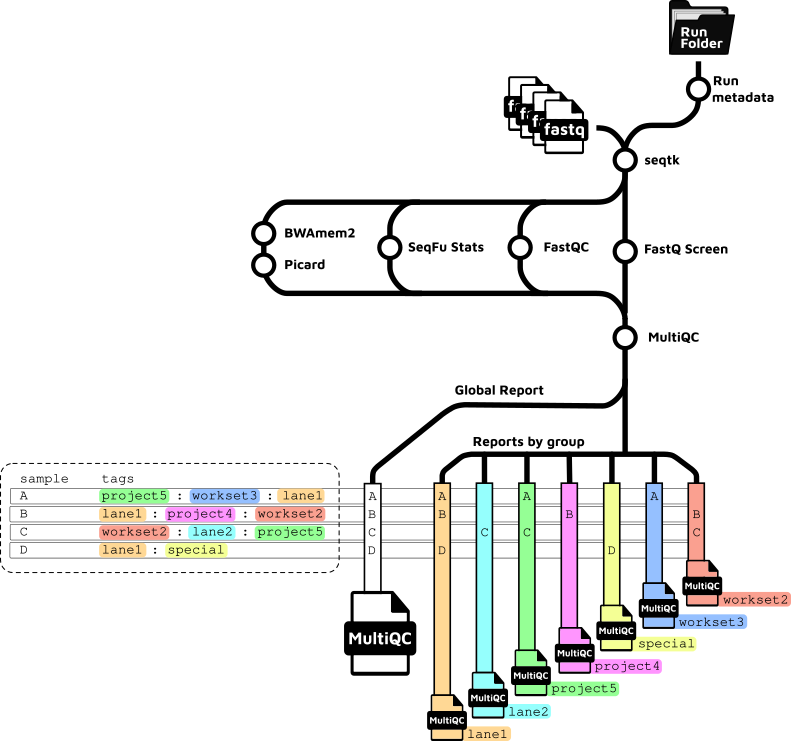
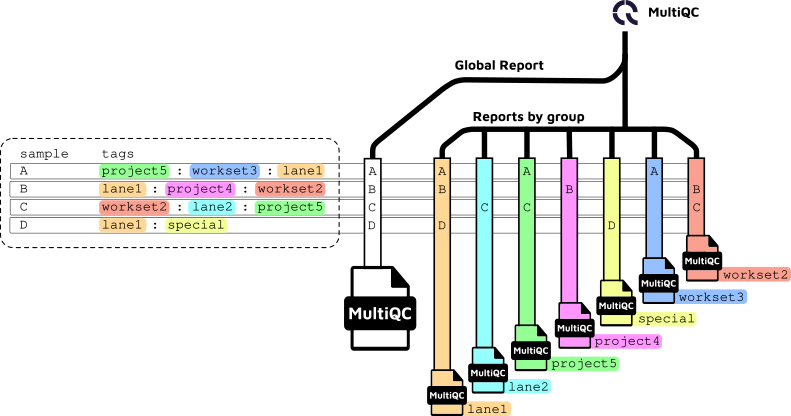
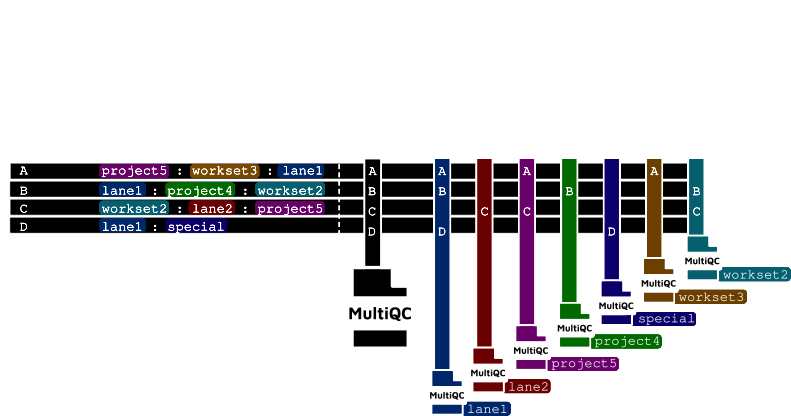
The diagram below illustrates the MultiQC reporting logic for per-sample tags and global aggregation.
Summary of tools and version used in the pipeline
| Tool | Version |
|---|---|
| bwamem2 | 2.3 |
| bbmap | 39.18 |
| checkQC | 4.1.0 |
| chelae | 0.1.0 |
| fq/lint | 0.12.0 |
| fastp | 1.1.0 |
| fastqc | 0.12.1 |
| fastqe | 0.5.2 |
| fastqscreen | 0.16.0 |
| kraken2 | 2.1.6 |
| krona | 2.8.1 |
| multiqc | 1.35 |
| multiqcsav | 0.2.0 |
| picard | 3.4.0 |
| riker | 0.4.0 |
| samtools | 1.24 |
| seqfu | 1.22.3 |
| seqkit | 2.9.0 |
| seqtk | 1.4 |
| sequali | 1.0.2 |
| toulligqc | 2.8.4 |
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,fastq_1,fastq_2,rundir,tagsCONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,200624_A00834_0183_BHMTFYDRXX,lane1:project5:group2Each row represents a fastq file (single-end with only fastq_1) or a pair of fastq files (paired end with fastq_1 and fastq_2).
rundir is the path to the runfolder.
tags is a colon-separated list of tags that will be added to the MultiQC report for this sample.
Now, you can run the pipeline using:
nextflow run nf-core/seqinspector \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/seqinspector was originally written by @agrima2010, @Aratz, @FranBonath, @kedhammar, and @MatthiasZepper from the Swedish National Genomics Infrastructure and Clinical Genomics Stockholm.
Maintenance is now lead by Maxime U Garcia (National Genomics Infrastructure)
We thank the following people for their extensive assistance in the development of this pipeline:
- @adamrtalbot
- @alneberg
- @beatrizsavinhas
- @ctuni
- @edmundmiller
- @EliottBo
- @erkutilaslan
- @KarNair
- @kjellinjonas
- @mahesh-panchal
- @matrulda
- @mirpedrol
- @nggvs
- @nkongenelly
- @Patricie34
- @pontushojer
- @ramprasadn
- @rannick
- @sarajeeeze
- @TMAdams
- @torigiffin
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #seqinspector channel (you can join with this invite).
Citations
You can cite the seqinspector zenodo record for a specific version using the following doi: 10.5281/zenodo.18757486
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.