2.2.4). The latest stable release is2.4.1.Introduction
nf-core/smrnaseq is a bioinformatics best-practice analysis pipeline used for small RNA sequencing data analysis.
This document describes the output produced by the pipeline. Most of the plots are taken from the MultiQC report, which summarises results at the end of the pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
- FastQC - read quality control
- FastP - adapter trimming
- Bowtie2 - contamination filtering
- Bowtie - alignment against mature miRNAs and miRNA precursors (hairpins)
- SAMtools - alignment result processing and feature counting
- edgeR - normalization, MDS plot and sample pairwise distance heatmap
- Bowtie - alignment against reference genome for QC purpose
- mirtop - miRNA and isomiR annotation
- miRDeep2 - known and novel miRNA annotation
- miRTrace - a comprehensive tool for QC purpose
- MultiQC - aggregate report, describing results of the whole pipeline
- Pipeline information - Report metrics generated during the workflow execution
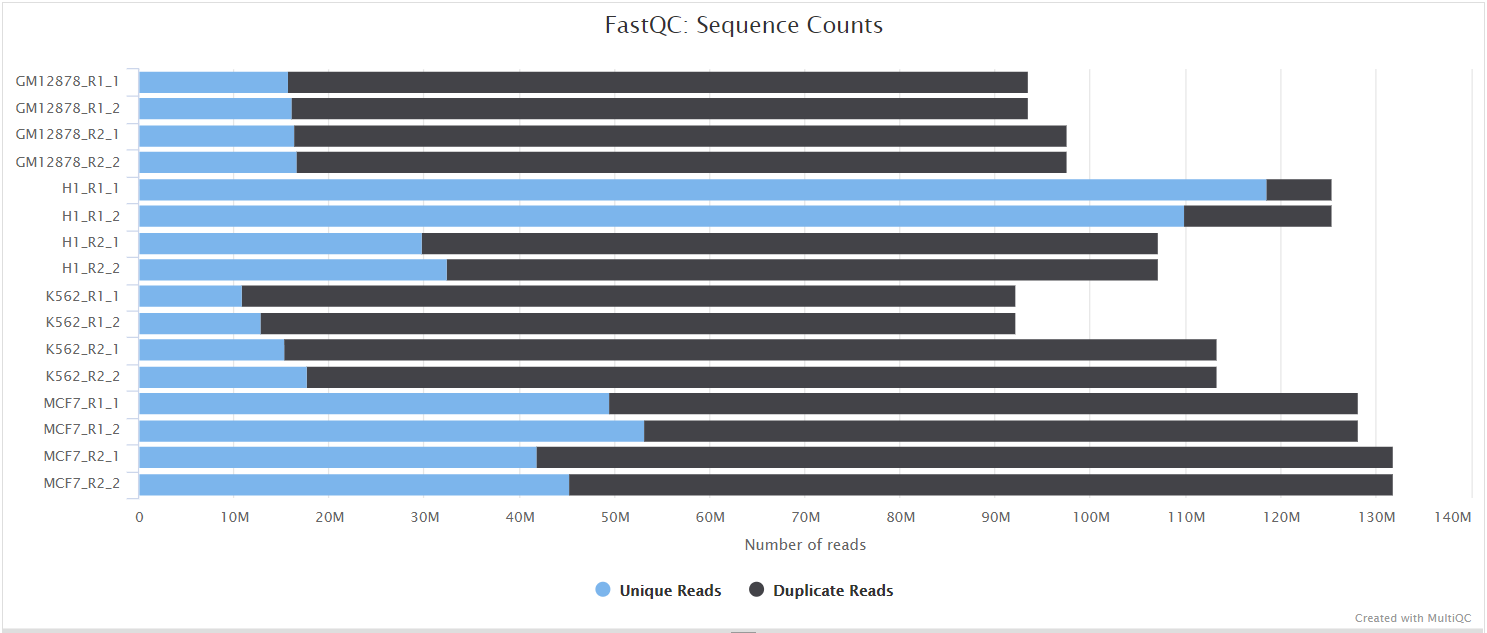
FastQC
Output files
fastqc/*_fastqc.html: FastQC report containing quality metrics.*_fastqc.zip: Zip archive containing the FastQC report, tab-delimited data file and plot images.
FastQC gives general quality metrics about your sequenced reads. It provides information about the quality score distribution across your reads, per base sequence content (%A/T/G/C), adapter contamination and overrepresented sequences. For further reading and documentation see the FastQC help pages.
FastP
FastP is used for removal of adapter contamination and trimming of low quality regions.
MultiQC reports the percentage of bases removed by FastP in the General Statistics table, along some further information on the results.
Output directory: results/fastp
Contains FastQ files with quality and adapter trimmed reads for each sample, along with a log file describing the trimming.
sample_fastp.json- JSON report file with information on parameters and trimming metricssample_fastp.html- HTML report with some visualizations of trimming metrics
FastP can automatically detect adapter sequences when not specified directly by the user - the pipeline also comes with a feature and a supplied miRNA adapters file to ensure adapters auto-detected are more accurate. If there are needs to add more known miRNA adapters to this list, please open a pull request.
Bowtie2
Bowtie2 is used to align the reads to user-defined databases of contaminants.
MultiQC reports the number of reads that were removed by each of the contaminant databases.
Bowtie
Bowtie is used for mapping adapter trimmed reads against the mature miRNAs and miRNA precursors (hairpins) of the chosen database miRBase or MirGeneDB.
Output directory: results/samtools
sample_mature.bam: The aligned BAM file of alignment against mature miRNAssample_mature_unmapped.fq.gz: Unmapped reads against mature miRNAs This file will be used as input for the alignment against miRNA precursors (hairpins)sample_mature_hairpin.bam: The aligned BAM file of alignment against miRNA precursors (hairpins) that didn’t map to the maturesample_mature_hairpin_unmapped.fq.gz: Unmapped reads against miRNA precursors (hairpins)sample_mature_hairpin_genome.bam: The aligned BAM file of alignment against that didn’t map to the precursor.
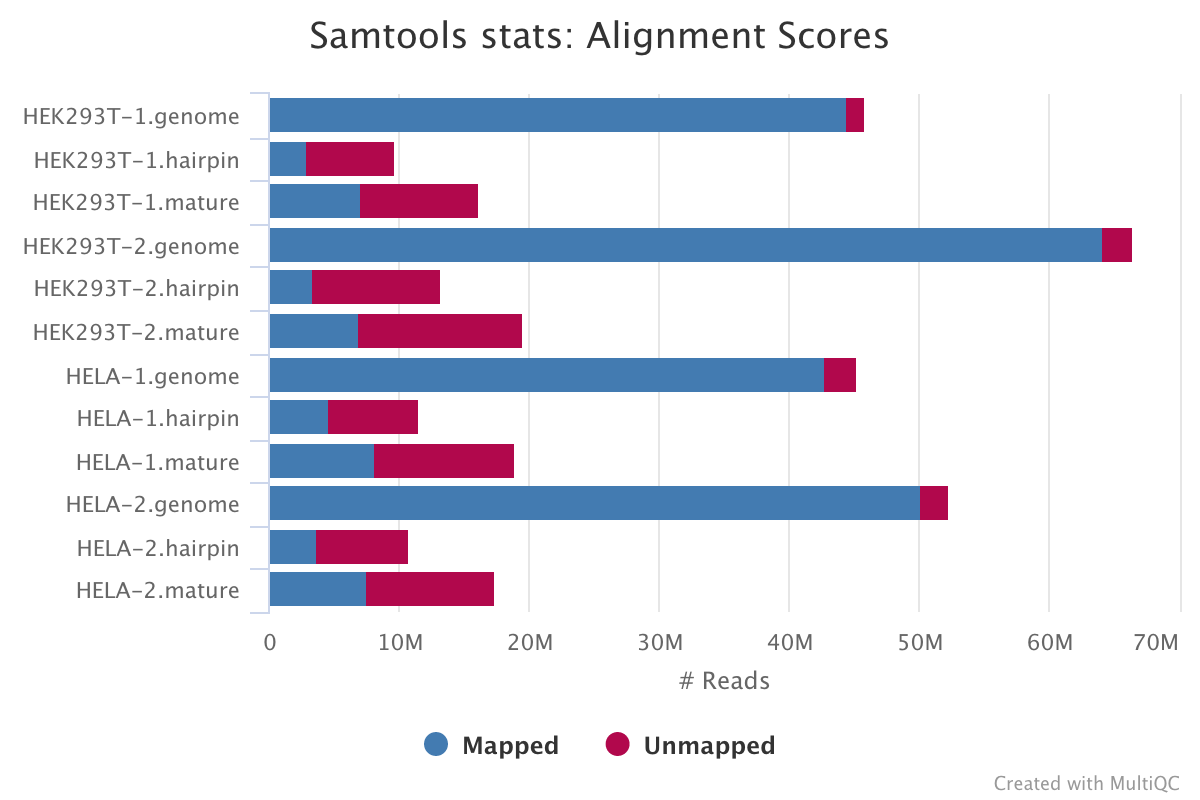
SAMtools
SAMtools is used for sorting and indexing the output BAM files from Bowtie. In addition, the numbers of features are counted with the idxstats option.
Output directory: results/samtools/samtools_stats
stats|idxstats|flagstat: BAM stats for each of the files listed above.
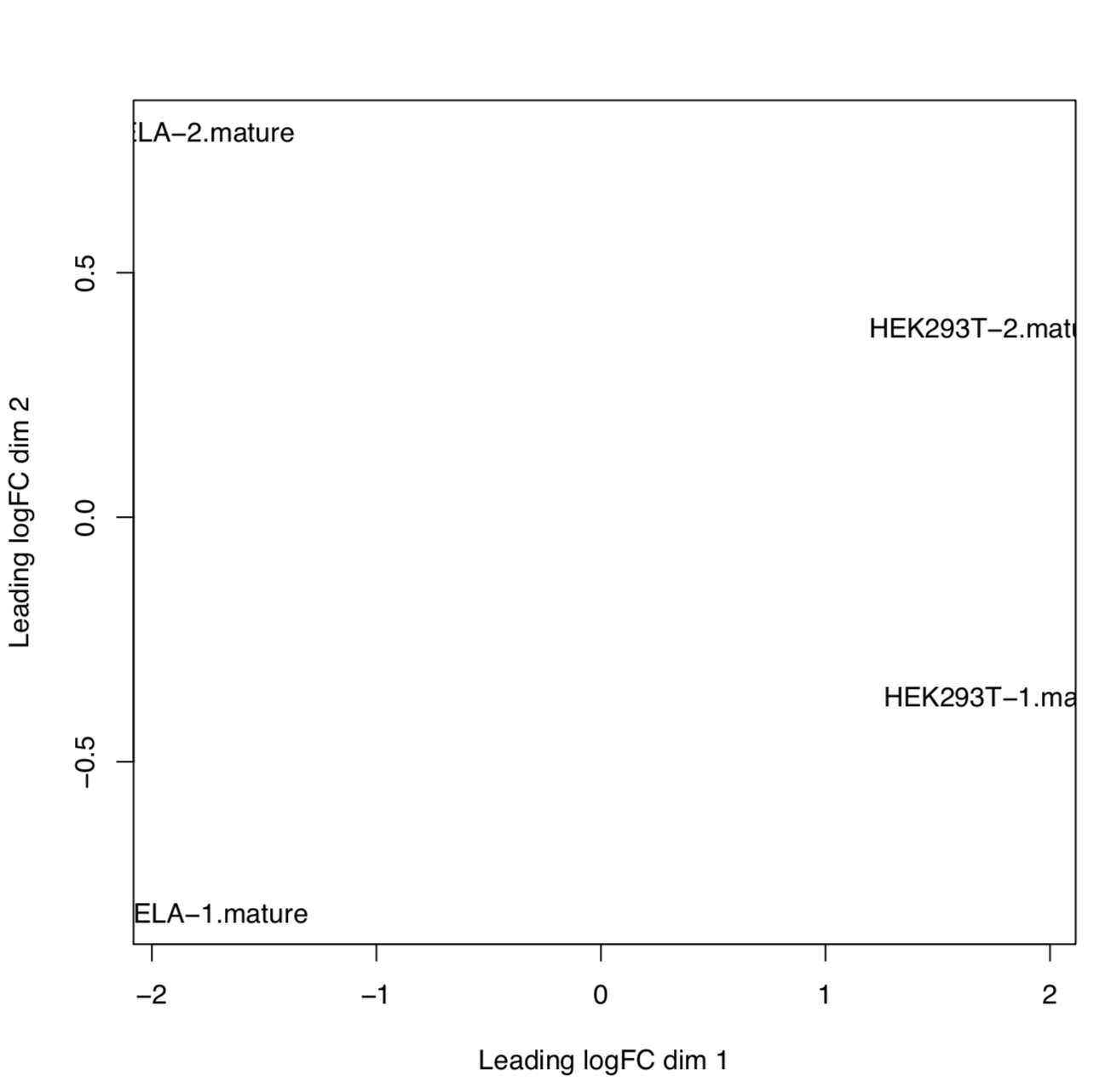
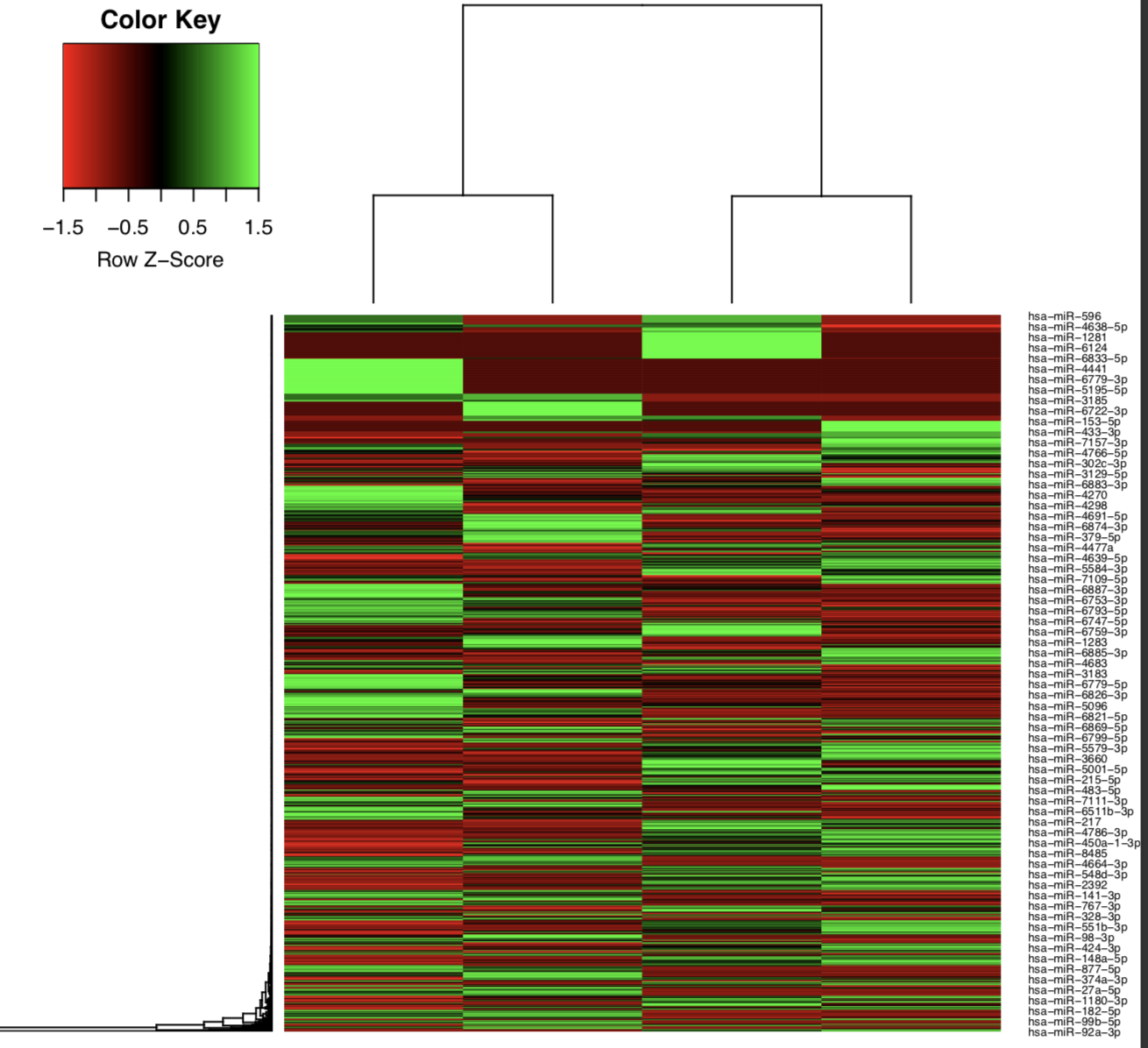
edgeR
edgeR is an R package used for differential expression analysis of RNA-seq expression profiles.
Output directory: results/edgeR
[mature/hairpin]_normalized_CPM.txtTMM normalized counts of reads aligned to mature miRNAs/miRNA precursors (hairpins)[mature/hairpin]_edgeR_MDS_plot.pdfMultidimensional scaling plot of all samples based on the expression profile of mature miRNAs/miRNA precursors (hairpins)[mature/hairpin]_CPM_heatmap.pdfHeatmap based on the expression profile of mature miRNAs/miRNA precursors (hairpins)[mature/hairpin]_log2CPM_sample_distances_dendrogram.pdfDendrograms of distance among samples based on the expression profile of mature miRNAs/miRNA precursors (hairpins)[mature/hairpin]_log2CPM_sample_distances_heatmap.pdfPairwise correlation of samples based on the expression profile of mature miRNAs/miRNA precursors (hairpins)
Example: MDS plot of 10 samples based on their expression profiles of mature miRNAs. Here we can see that samples cluster based on different sample types and library preparation kits.
Example: Heatmap of tumor and normal samples based on the top differentially expressed mature miRNAs.
mirtop
mirtop is used to parse the BAM files from bowtie alignment, and produce a mirgff3 file with information about miRNAs and isomirs.
Output directory: results/mirtop
mirtop.gff: mirgff3 filemirtop.tsv: tabular file of the previous file for easy integration with downstream analysis.mirtop_rawData.tsv: File compatible with isomiRs Bioconductor package to perform isomiRs analysis.mirna.tsv: tabular file with miRNA counts after summarizing unique isomiRs for each miRNA
miRDeep2
miRDeep2 is used for the identification of novel and known miRNAs in deep sequencing data.
Output directory: results/mirdeep2
mirdeep/timestamp_sample.bedFile with the known and novel miRNAs in bed format.mirdeep/timestamp_sample.csvFile with an overview of all detected miRNAs (known and novel) in csv format.mirdeep/timestamp_sample.htmlA HTML report with an overview of all detected miRNAs (known and novel) in html format.
miRTrace
miRTrace is a quality control specifically for small RNA sequencing data (smRNA-Seq). Each sample is characterized by profiling sequencing quality, read length, sequencing depth and miRNA complexity and also the amounts of miRNAs versus undesirable sequences (derived from tRNAs, rRNAs and sequencing artifacts).
Output directory: results/mirtrace
mirtrace-report.htmlAn interactive HTML report summarizing all output statistics from miRTracemirtrace-results.jsonA JSON file with all output statistics from miRTracemirtrace-stats-*.tsvTab-separated statistics filesqc_passed_reads.all.collapsedFASTA file per sample with sequence reads that passed QC in miRTraceqc_passed_reads.rnatype_unknown.collapsedFASTA file per sample with unknown reads in the RNA type analysis
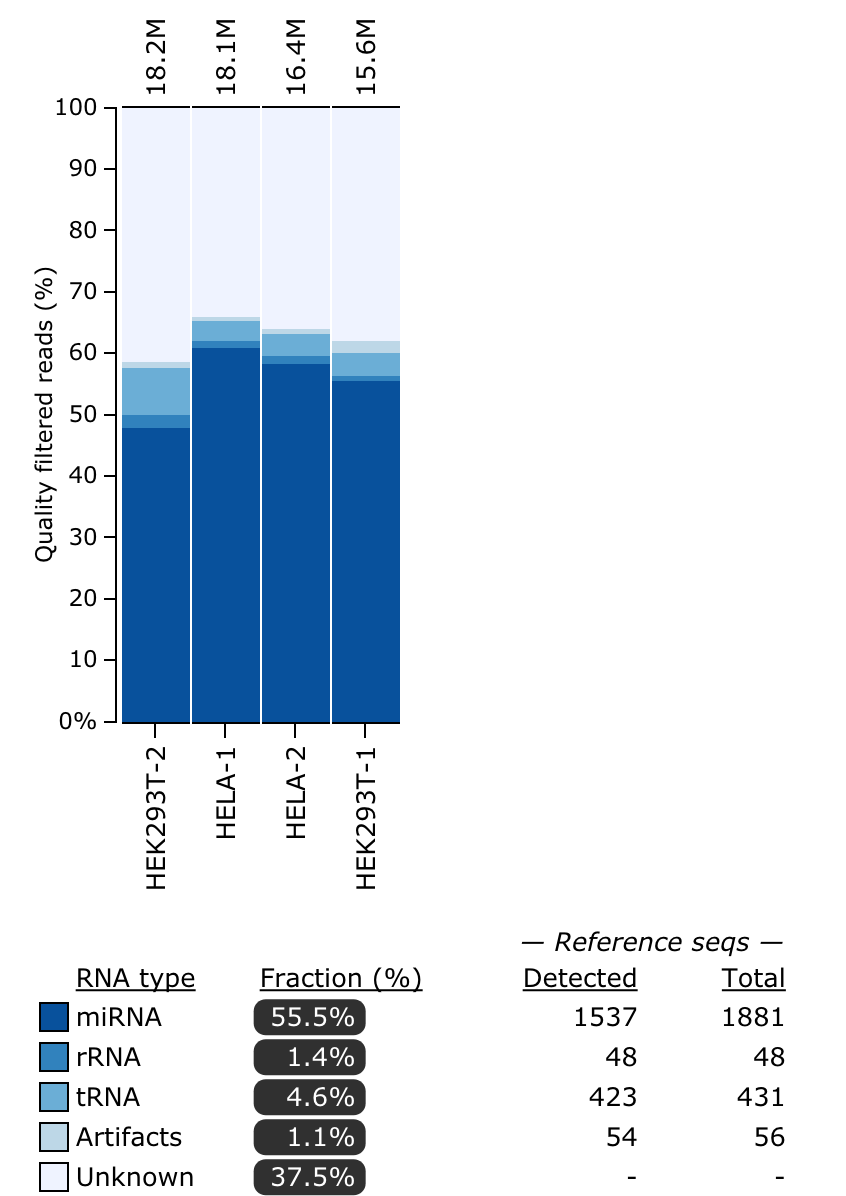
Refer to the tool manual for detailed specifications about output files. Here is an example of the RNA types plot that you will see:
MultiQC
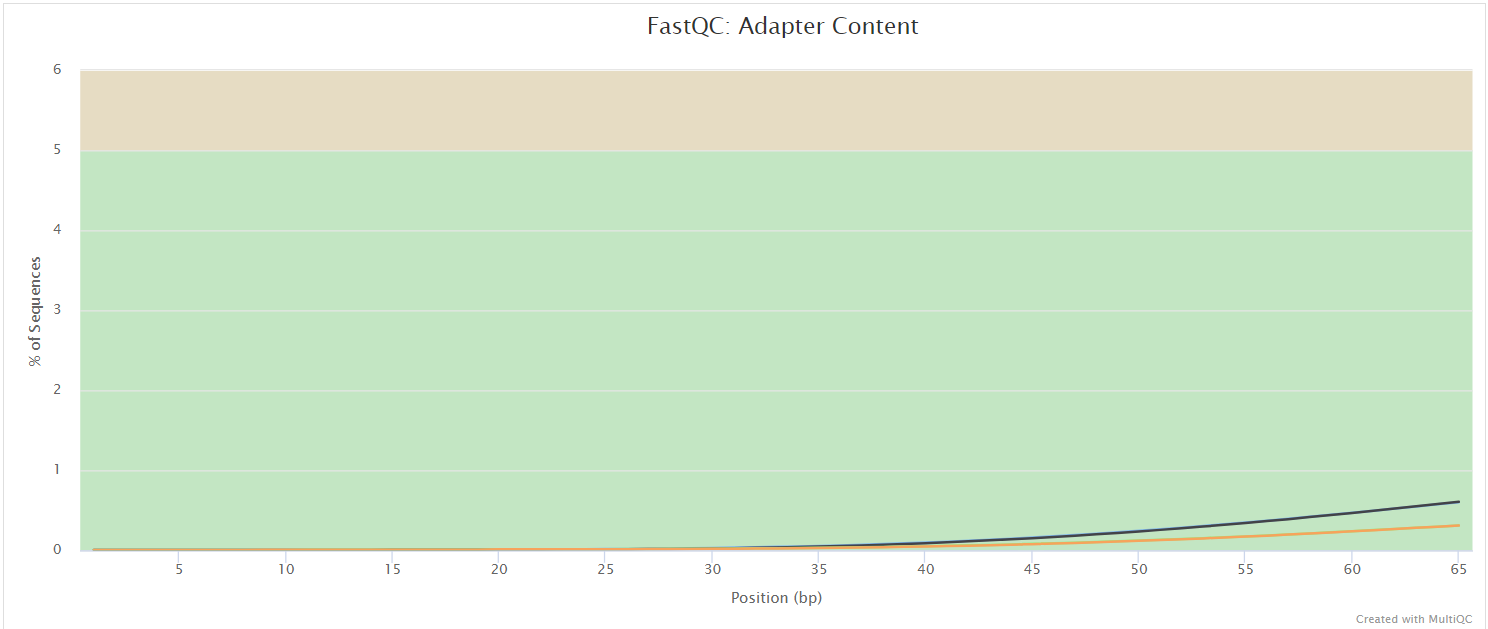
The FastQC plots displayed in the MultiQC report shows untrimmed reads. They may contain adapter sequence and potentially regions with low quality.
MultiQC
Output files
multiqc/multiqc_report.html: a standalone HTML file that can be viewed in your web browser.multiqc_data/: directory containing parsed statistics from the different tools used in the pipeline.multiqc_plots/: directory containing static images from the report in various formats.
MultiQC is a visualization tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available in the report data directory.
Results generated by MultiQC collate pipeline QC from supported tools e.g. FastQC. The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability. For more information about how to use MultiQC reports, see http://multiqc.info.
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter’s are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv. - Parameters used by the pipeline run:
params.json.
- Reports generated by Nextflow:
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.