nf-core/spatialxe
Introduction
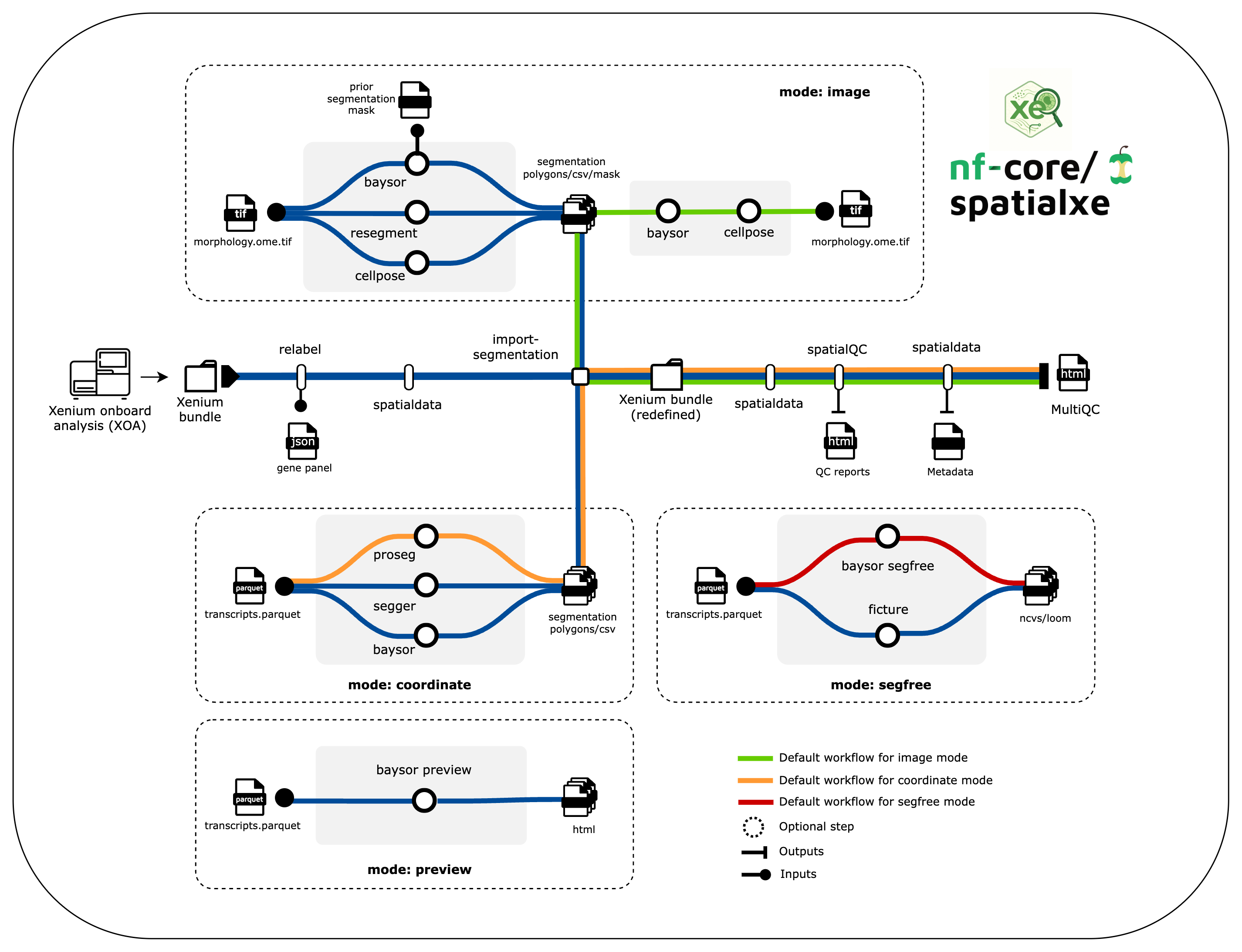
nf-core/spatialxe is a bioinformatics best-practice processing and quality control pipeline for Xenium data. The pipeline is currently under developement and not completed yet!. The current plan for the pipeline implementation is shown in the metromap below. Please note that the pipeline steps and methods might change as we move forward in the development cycle.
Usage
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Pipeline summary
Quick Start
samplesheet.csv:
sample,bundle,image
test_sample,/path/to/xenium-bundle,/path/to/morphology.ome.tifNow, you can run the pipeline using:
Run image-based segmentation mode
CELLPOSE -> BAYSOR -> XR-IMPORT_SEGMENTATION -> SPATIALDATA -> QC
nextflow run nf-core/spatialxe \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR> \
--mode <MODE>Run coordinate-based segmentation mode
PROSEG -> PROSEG2BAYSOR -> XR-IMPORT_SEGMENTATION -> SPATIALDATA -> QC
nextflow run nf-core/spatialxe \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR> \
--mode coordinateRun segfree mode
BAYSOR_SEGFREE
nextflow run nf-core/spatialxe \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR> \
--mode segfreeRun preview mode
BAYSOR_PREVIEW
nextflow run nf-core/spatialxe \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR> \
--mode previewPlease provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Runtime and resource estimations
| Tool | Compute | Runtime (min / med / max) | Peak RSS (min / med / max) |
|---|---|---|---|
| Cellpose | GPU | 1m / 4m / 1.4h | 10 GB / 26 GB / 554 GB |
| Cellpose | CPU | 1.3h / 2.3h / 6.5h | 161 GB / 426 GB / 1115 GB |
| StarDist | GPU | 1m / 4m / 7m | 5 GB / 12 GB / 18 GB |
| StarDist | CPU | 5m / 6m / 7m | 18 GB / 18 GB / 18 GB |
| Segger (create_dataset) | GPU | 2m / 9m / 31m | 1.7 GB / 14 GB / 50 GB |
| Segger (create_dataset) | CPU | 13m / 21m / 46m | 13 GB / 19 GB / 49 GB |
| Segger (train) | GPU | 10m / 43m / 2.9h | 30 GB / 33 GB / 60 GB |
| Segger (predict) | GPU | 2m / 16m / 59m | 10 GB / 25 GB / 87 GB |
| Baysor (whole-image) | CPU | 2m / 30m / 17h | 6 GB / 10 GB / 650 GB |
| Baysor (tiled) | CPU | 1m / 18m / 13h | 0.2 GB / 34 GB / 530 GB |
| Proseg | CPU | 1m / 18m / 6.8h | 279 MB / 3.8 GB / 136 GB |
| XeniumRanger (resegment) | CPU | 18m / 39m / 3.7h | 28 GB / 54 GB / 60 GB |
| XeniumRanger (import_seg) | CPU | 2m / 7m / 2.7h | 2.6 GB / 11 GB / 51 GB |
| Ficture (preprocess) | CPU | 3m / 4m / 13m | 331 MB / 357 MB / 21 GB |
- Cellpose GPU vs CPU: 35x faster on GPU (4m median vs 2.3h), 16x less memory (26 GB vs 426 GB)
- Segger: Only tool that truly requires GPU for all 3 steps (create_dataset, train, predict)
- StarDist: Very fast on CPU, GPU is not necessary to run its default model
Credits
nf-core/spatialxe is mainly developed by Sameesh Kher, Dongze He, and Florian Heyl.
We thank the following people for their extensive assistance in the development of this pipeline:
- Tobias Krause
- Krešimir Beštak (kbestak)
- Matthias Hörtenhuber (mashehu)
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #spatialxe channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.