nf-core/airrflow
B-cell and T-cell Adaptive Immune Receptor Repertoire (AIRR) sequencing analysis pipeline using the Immcantation framework
Introduction
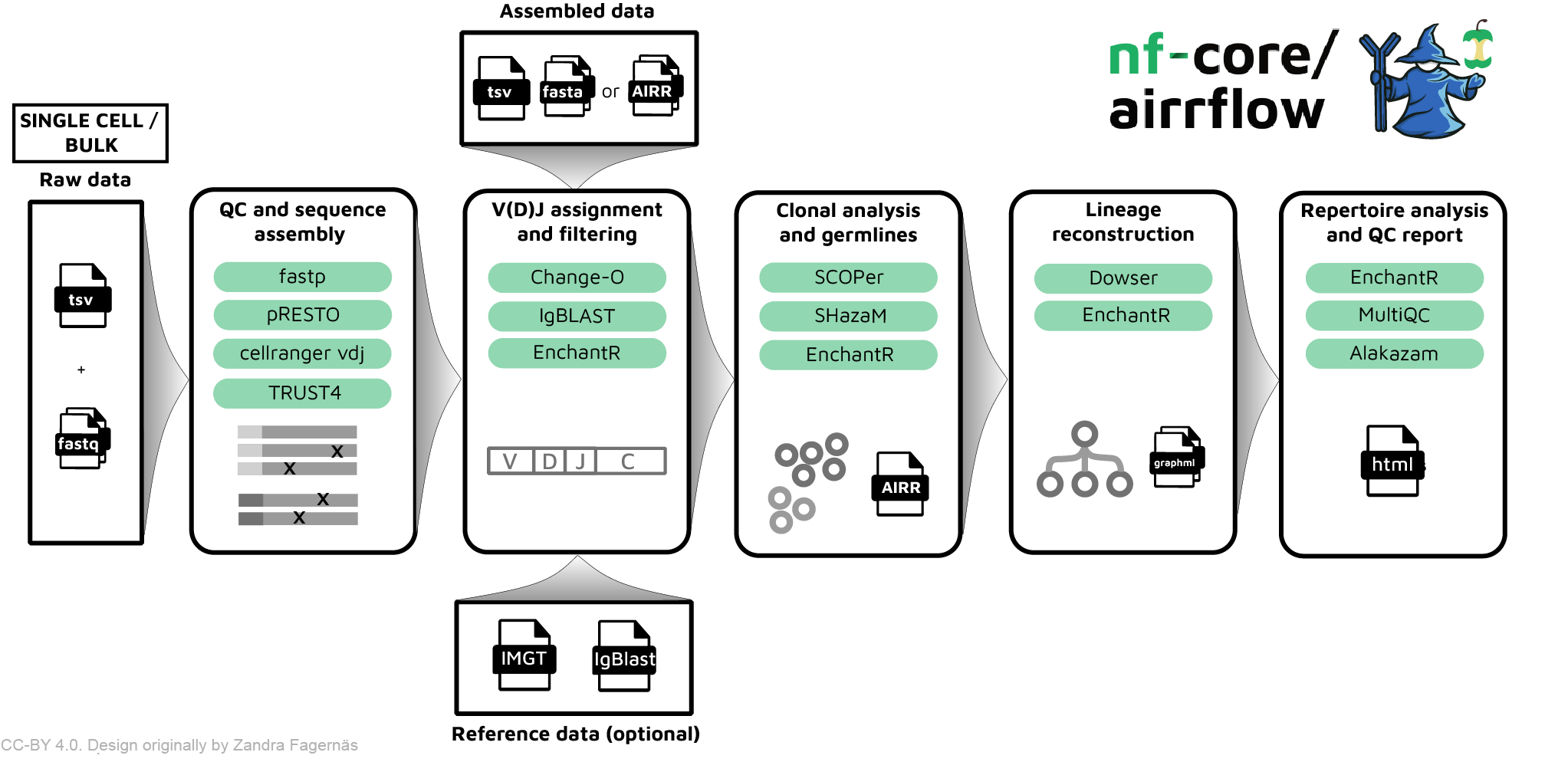
nf-core/airrflow is a bioinformatics best-practice pipeline to analyze B-cell receptor (BCR) or T-cell receptor (TCR) repertoire sequencing data. It allows the processing of targeted bulk and single-cell adaptive immune receptor sequencing data (AIRR-seq), as well as the extraction of TCR and BCR sequences from untargeted bulk and single-cell RNA-seq data. The pipeline enables and end-to-end analysis, departing from raw reads or readily assembled sequences, and performs sequence assembly, V(D)J assignment, clonal group inference, lineage reconstruction and repertoire analysis using the Immcantation framework, as well as other immune repertoire analysis tools.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where possible, these processes have been submitted to and installed from nf-core/modules in order to make them available to all nf-core pipelines, and to everyone within the Nextflow community!
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Pipeline summary
nf-core/airrflow allows the end-to-end processing of BCR and TCR bulk and single cell targeted sequencing data, as well as extracting BCR and TCR sequences from bulk and single-cell RNA-seq datasets. Several protocols are supported, please see the usage documentation for more details on the supported protocols. The pipeline has been certified as AIRR compliant by the AIRR community, which means that it is compatible with downstream analysis tools also supporting this format.
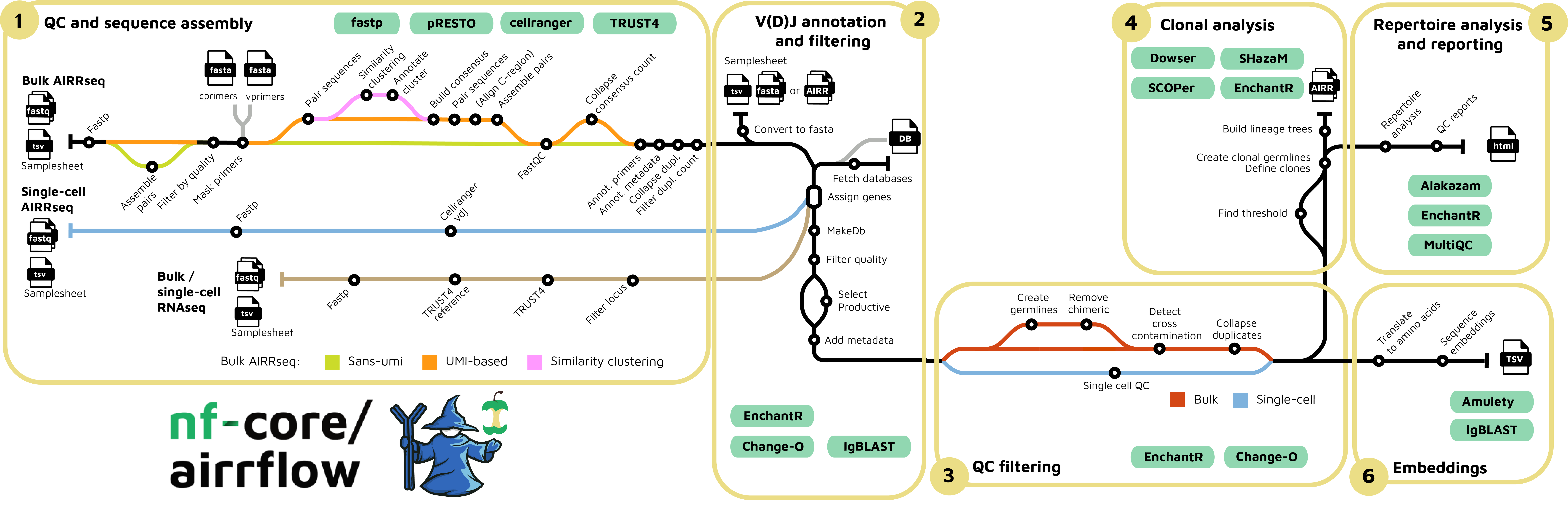
- QC and sequence assembly
- Bulk
- Raw read quality control, adapter trimming and clipping (
Fastp). - Filter sequences by base quality (
pRESTO FilterSeq). - Mask amplicon primers (
pRESTO MaskPrimers). - Pair read mates (
pRESTO PairSeq). - For UMI-based sequencing:
- Cluster sequences according to similarity (optional for insufficient UMI diversity) (
pRESTO ClusterSets). - Build consensus of sequences with the same UMI barcode (
pRESTO BuildConsensus).
- Cluster sequences according to similarity (optional for insufficient UMI diversity) (
- Assemble R1 and R2 read mates (
pRESTO AssemblePairs). - Remove and annotate read duplicates (
pRESTO CollapseSeq). - Filter out sequences that do not have at least 2 duplicates (
pRESTO SplitSeq).
- Raw read quality control, adapter trimming and clipping (
- Single cell
- cellranger vdj
- Assemble contigs
- Annotate contigs
- Call cells
- Generate clonotypes
- cellranger vdj
- V(D)J annotation and filtering (bulk and single-cell)
- Assign gene segments with
IgBlastusing a germline reference (Change-O AssignGenes). - Annotate alignments in AIRR format (
Change-O MakeDB) - Filter by alignment quality (locus matching v_call chain, min 200 informative positions, max 10% N nucleotides)
- Filter productive sequences (
Change-O ParseDB split) - Filter junction length multiple of 3
- Annotate metadata (
EnchantR)
- QC filtering (bulk and single-cell)
- Bulk sequencing filtering:
- Remove chimeric sequences (optional) (
SHazaM,EnchantR) - Detect cross-contamination (optional) (
EnchantR) - Collapse duplicates (
Alakazam,EnchantR)
- Remove chimeric sequences (optional) (
- Single-cell QC filtering (
EnchantR)- Remove cells without heavy chains.
- Remove cells with multiple heavy chains.
- Remove sequences in different samples that share the same
cell_idand nucleotide sequence, and thus are very likely contaminants. - Modify
cell_ids to ensure they are unique in each run.
- Clonal analysis (bulk and single-cell)
- Find threshold for clone definition (
SHazaM,EnchantR). - Create germlines and define clones, repertoire analysis (
SCOPer,EnchantR). - Build lineage trees (
Dowser,IgphyML,RAxML,EnchantR).
- Repertoire analysis and reporting
- Custom repertoire analysis pipeline report (
Alakazam). - Aggregate QC reports (
MultiQC).
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. You will also need to install a container engine such as Docker or Apptainer - formerly singularity - prior to running the pipeline. Make sure to test your setup with -profile test before running the workflow on actual data.
First, ensure that the pipeline tests run on your infrastructure:
nextflow run nf-core/airrflow -profile test,<docker/singularity/apptainer/podman/shifter/charliecloud/conda/institute> --outdir <OUTDIR>To run nf-core/airrflow with your data, you will need to first prepare a tab-separated samplesheet with the paths to your input data and necessary metadata to run the analysis. Depending on the input data type (bulk or single-cell, raw reads or assembled reads) the input samplesheet will vary. Please follow the documentation on samplesheets for more details. An example samplesheet for running the pipeline on bulk BCR / TCR sequencing data departing from raw reads looks as follows:
| sample_id | filename_R1 | filename_R2 | filename_I1 | subject_id | species | pcr_target_locus | tissue | sex | age | biomaterial_provider | single_cell | intervention | collection_time_point_relative | cell_subset |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample01 | sample1_S8_L001_R1_001.fastq.gz | sample1_S8_L001_R2_001.fastq.gz | sample1_S8_L001_I1_001.fastq.gz | Subject02 | human | IG | blood | NA | 53 | sequencing_facility | FALSE | Drug_treatment | Baseline | plasmablasts |
| sample02 | sample2_S8_L001_R1_001.fastq.gz | sample2_S8_L001_R2_001.fastq.gz | sample2_S8_L001_I1_001.fastq.gz | Subject02 | human | IG | blood | female | 78 | sequencing_facility | FALSE | Drug_treatment | Baseline | plasmablasts |
Each row represents a sample with fastq files (paired-end).
A typical command to run the pipeline from bulk raw fastq files is:
nextflow run nf-core/airrflow \
-r <release> \
-profile <docker/singularity/podman/shifter/charliecloud/conda/institute> \
--mode fastq \
--input input_samplesheet.tsv \
--library_generation_method specific_pcr_umi \
--cprimers CPrimers.fasta \
--vprimers VPrimers.fasta \
--umi_length 12 \
--umi_position R1 \
--outdir ./resultsFor common bulk sequencing protocols we provide pre-set profiles that specify primers, UMI length, etc for common commercially available sequencing protocols. Please check the Supported protocol profiles for a full list of available profiles. An example command running the NEBNext UMI protocol profile with docker containers is:
nextflow run nf-core/airrflow \
-profile nebnext_umi,docker \
--mode fastq \
--input input_samplesheet.tsv \
--outdir resultsA typical command to run the pipeline from single cell raw fastq files (10X genomics) is:
nextflow run nf-core/airrflow -r dev \
-profile <docker/singularity/podman/shifter/charliecloud/conda/institute> \
--mode fastq \
--input input_samplesheet.tsv \
--library_generation_method sc_10x_genomics \
--reference_10x reference/refdata-cellranger-vdj-GRCh38-alts-ensembl-5.0.0.tar.gz \
--outdir ./resultsA typical command to run the pipeline from single-cell AIRR rearrangement tables or assembled bulk sequencing fasta data is:
nextflow run nf-core/airrflow \
-r <release> \
-profile <docker/singularity/podman/shifter/charliecloud/conda/institute> \
--input input_samplesheet.tsv \
--mode assembled \
--outdir resultsSee the usage documentation and the parameter documentation for more details on how to use the pipeline and all the available parameters.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those
provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the the results of a test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/airrflow was originally written by:
We thank the following people for their extensive assistance in the development of the pipeline:
Acknowledgements
![]()
The nf-core/airrflow pipeline development is partly supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) grant U01AI184647 to Gisela Gabernet.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #airrflow channel (you can join with this invite).
Citations
If you use nf-core/airrflow for your analysis, please cite the article as follows:
nf-core/airrflow: an adaptive immune receptor repertoire analysis workflow employing the Immcantation framework
Gisela Gabernet, Susanna Marquez, Robert Bjornson, Alexander Peltzer, Hailong Meng, Edel Aron, Noah Y. Lee, Cole G. Jensen, David Ladd, Mark Polster, Friederike Hanssen, Simon Heumos, nf-core community, Gur Yaari, Markus C. Kowarik, Sven Nahnsen, Steven H. Kleinstein. (2024) PLOS Computational Biology, 20(7), e1012265. doi: https://doi.org/10.1371/journal.pcbi.1012265. Pubmed PMID: 39058741.
The specific pipeline version using the following DOI: 10.5281/zenodo.2642009
Please also cite all the tools that are being used by the pipeline. An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.