Introduction
nf-core/callingcards is a bioinformatics pipeline that can be used to process raw Calling Cards data obtained from either mammals (human, mouse) or yeast. It takes a samplesheet, which describes the sample names, paths to the fastq files, and paths to the barcode details json files, a parameter which identifies the
organism type (either mammals or yeast) and either the name of the reference genome on igenomes or paths to the fasta/gff. It then parses the reads, counts the
number of calling cards insertions, and provides some QC metrics.
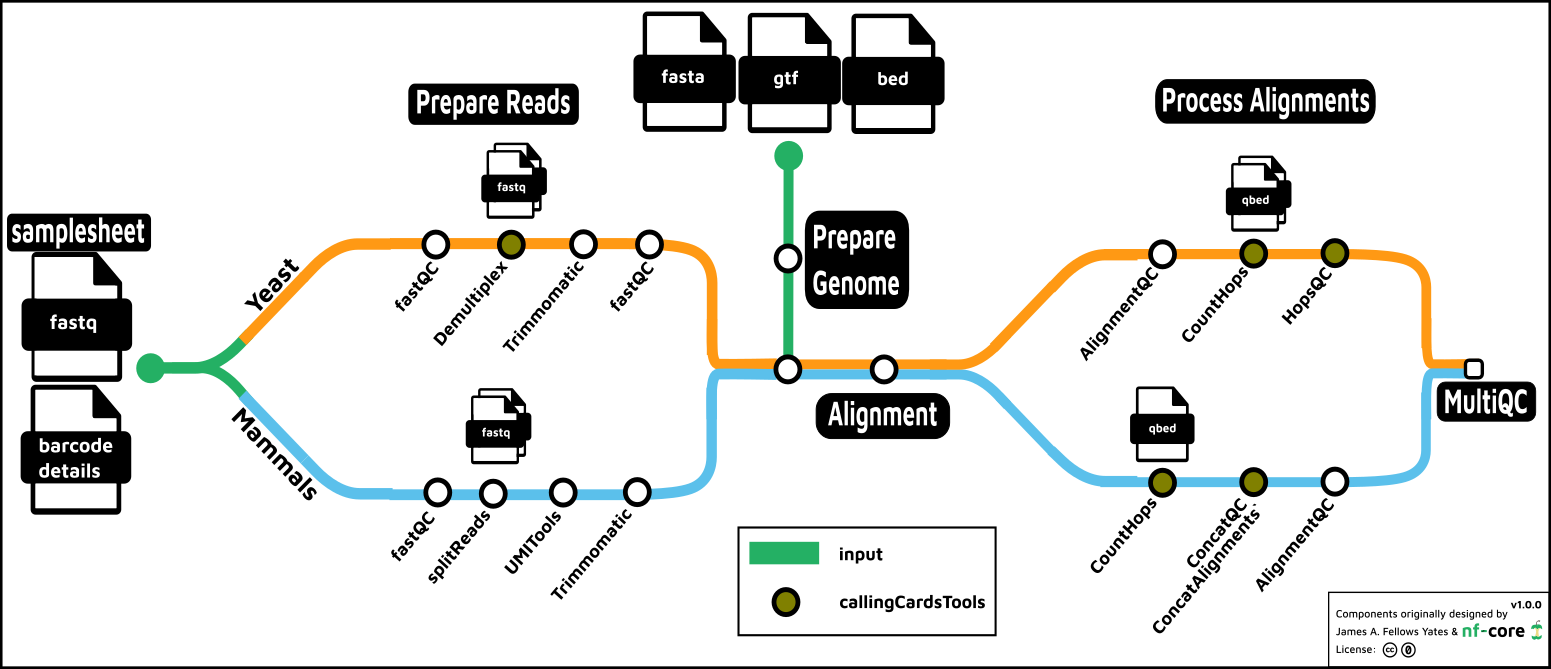
- Prepare the Genome
- Prepare the Reads
- Read QC (
FastQC) - Split reads
- Yeast: demultiplex by barcode (
callingCardsTools) - Mammals: split for parallel processing (
seqkit)- Mammals: extract barcode to fastq read ID(
UMItools)
- Mammals: extract barcode to fastq read ID(
- Yeast: demultiplex by barcode (
- Optionally trim reads (
Trimmomatic)])
- Read QC (
- Align
- Count Hops (
callingCardsTools) - Present QC data (
MultiQC)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how
to set-up Nextflow. Make sure to test your setup
with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data according to your organism.
A Yeast samplesheet will look like:
yeast_samplesheet.csv:
sample,fastq_1,fastq_2,barcode_details
run_6177,run_6177_sample_R1.fastq.gz,run_6177_sample_R2.fastq.gz,run_6177_barcode_details.jsonEach row represents a multiplexed fastq file where the barcode_details.json file describes the barcodes which correspond to each transcription factor in the library.
A mammals (human, mouse) samplesheet will look like:
mammals_samplesheet.csv:
sample,fastq_1,fastq_2,barcode_details
midbrain_rep3,midbrain_rep3_R1.fastq.gz,,barcode_details.jsonNote that currently, the mammals workflow expects only R1 reads.
Now, you can run the pipeline using:
nextflow run nf-core/callingcards \
-profile <docker/singularity/.../institute>,<default_yeast/default_mammals> \
--input samplesheet.csv \
--genome <igenomes_name (only required for mammals)> \
--outdir <OUTDIR>Note that the default_yeast and default_mammals profiles are provided for convenience. You should check the parameters which these set to ensure that they are what you need for your data.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/callingcards is implemented in nextflow by Chase Mateusiak. It was adapted from scripts written by:
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #callingcards channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.