Introduction
nf-core/dartseq is a workflow for DART-seq style RNA sequencing analyses with optional RNA editing downstream steps. It accepts single-end or paired-end FASTQ input, performs read QC and alignment, and can run Bullseye- and RustQC-based post-processing. Standard outputs include per-sample QC reports, alignments, MultiQC summaries, and optional edited-site tables.
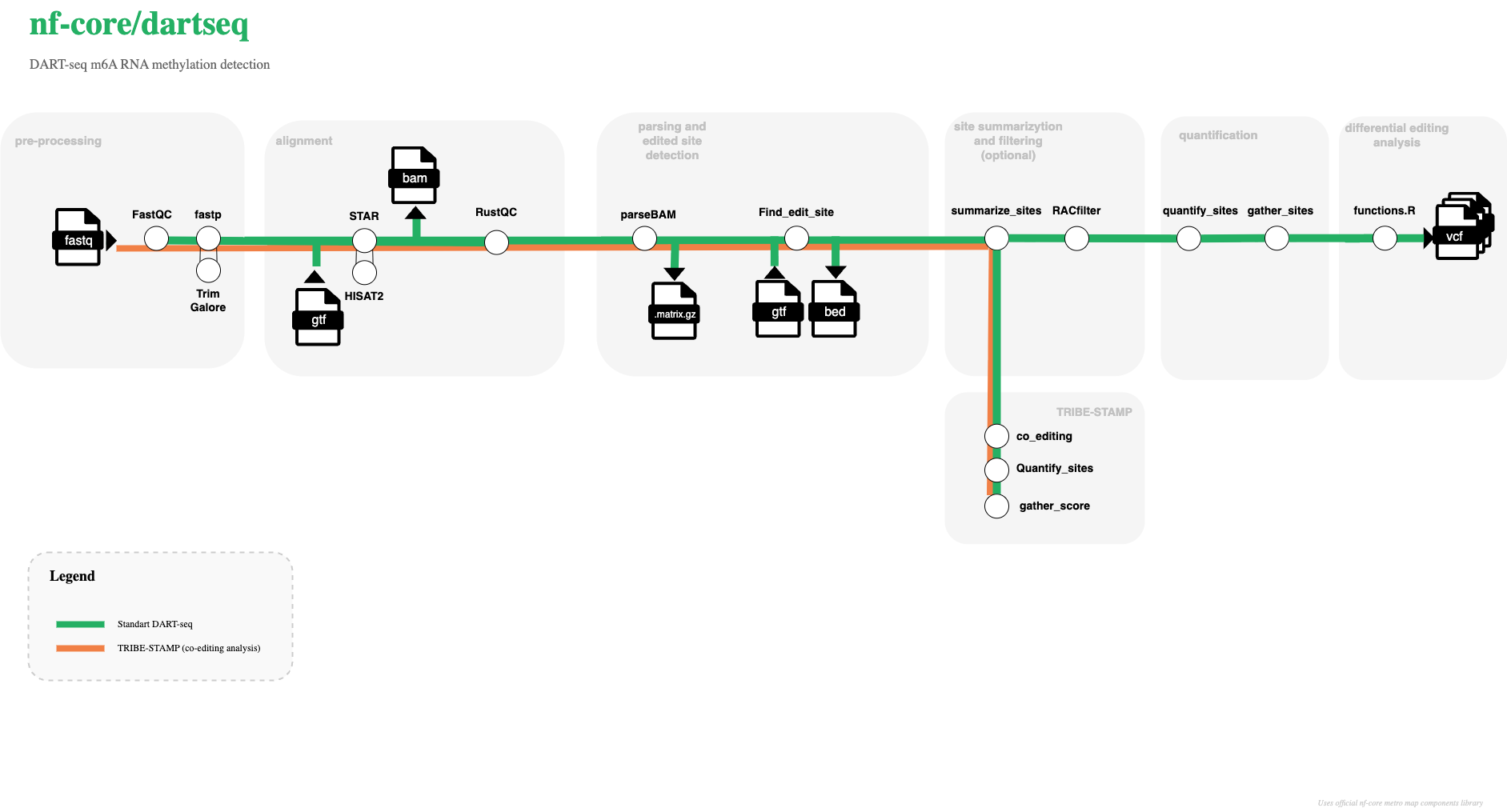
Workflow overview:
- Parse and validate the input samplesheet.
- Trim reads with
fastporTrim Galore(or skip trimming if requested). - Run per-sample quality control with FastQC.
- Build or use provided aligner references (STAR or HISAT2).
- Align reads and produce sorted BAM files plus index files.
- Run optional Bullseye editing analysis: parse BAM, summarize sites, quantify edits, compare against controls, and optionally RAC filter / gather sites / GLM.
- Run optional RustQC summaries on alignments.
- Aggregate run-wide summaries and software versions with MultiQC.
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Prepare a samplesheet with at least sample, fastq_1, and fastq_2 columns.
For single-end data, leave fastq_2 empty.
Now, you can run the pipeline using:
nextflow run nf-core/dartseq \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/dartseq was originally written by Mathieu Flamand, Olga Brovkina, Joana Pimenta Bernardes, Fatemeh Nasehi.
We thank the following people for their extensive assistance in the development of this pipeline:
- The nf-core maintainers and community contributors who helped evolve the template integration and testing strategy.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #dartseq channel (you can join with this invite).
Citations
If this pipeline is used in a publication, cite the nf-core framework below and the software references in CITATIONS.md.
After the first release, update the placeholder DOI 10.5281/zenodo.XXXXXXX in this README with the pipeline Zenodo DOI.
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.