nf-core/demultiplex
Demultiplexing pipeline for sequencing data
1.4.0). The latest
stable release is
1.7.0
.
Introduction
nf-core/demultiplex is a bioinformatics pipeline used to demultiplex the raw data produced by next generation sequencing machines. The following platforms are supported:
- Illumina (via
bcl2fastqorbclconvert) - Element Biosciences (via
bases2fastq) - Singular Genomics (via
sgdemux) - FASTQ files with user supplied read structures (via
fqtk)
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where possible, these processes have been submitted to and installed from nf-core/modules in order to make them available to all nf-core pipelines, and to everyone within the Nextflow community!
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources.The results obtained from the full-sized test can be viewed on the nf-core website.
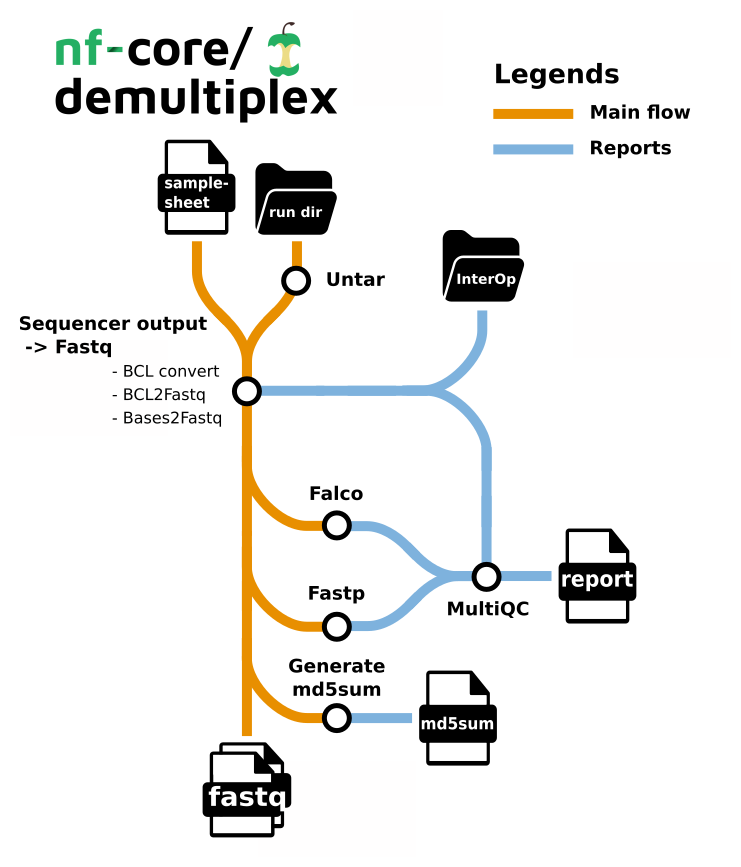
Pipeline summary
- Demultiplexing
- bcl-convert - converting bcl files to fastq, and demultiplexing (CONDITIONAL)
- bases2fastq - converting bases files to fastq, and demultiplexing (CONDITIONAL)
- bcl2fastq - converting bcl files to fastq, and demultiplexing (CONDITIONAL)
- sgdemux - demultiplexing bgzipped fastq files produced by Singular Genomics (CONDITIONAL)
- fqtk - a toolkit for working with FASTQ files, written in Rust (CONDITIONAL)
- fastp - Adapter and quality trimming
- Falco - Raw read QC
- md5sum - Creates an MD5 (128-bit) checksum of every fastq.
- MultiQC - aggregate report, describing results of the whole pipeline
Usage
If you are new to Nextflow and nf-core, please refer to this page on how
to set-up Nextflow. Make sure to test your setup
with -profile test before running the workflow on actual data.
nextflow run nf-core/demultiplex --input samplesheet.csv --outdir <OUTDIR> -profile <docker/singularity/podman/shifter/charliecloud/conda/institute>nextflow run nf-core/demultiplex \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those
provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
The nf-core/demultiplex pipeline was written by Chelsea Sawyer from The Bioinformatics & Biostatistics Group for use at The Francis Crick Institute, London.
The pipeline was re-written in Nextflow DSL2 and is primarily maintained by Matthias De Smet(@matthdsm) from Center For Medical Genetics Ghent, Ghent University and Edmund Miller(@emiller88) from Element Biosciences
We thank the following people for their extensive assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #demultiplex channel (you can join with this invite).
Citations
If you use nf-core/demultiplex for your analysis, please cite it using the following doi: 10.5281/zenodo.7153103
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.