nf-core/diaproteomics
Automated quantitative analysis of DIA proteomics mass spectrometry measurements.
22.10.6.Learn more.Introduction
nfcore/diaproteomics is a bioinformatics analysis pipeline used for quantitative processing of data independant (DIA) proteomics data (preprint available here).
The workflow is based on the OpenSwathWorkflow for SWATH-MS proteomic data. DIA RAW files (mzML) serve as inputs and library search is performed based on a given input spectral library. Optionally, spectral libraries can be generated (EasyPQP) from multiple matched DDA measurments and respective search results. Generated libraries can then further be aligned applying a pairwise RT alignment and concatenated into a single large library. In the same way internal retention time standarts (irts) can be either supplied or generted by the workflow in order to align library and DIA measurements into the same retention time space. FDR rescoring is applied using Pyprophet based on a competitive target-decoy approach on peakgroup or global peptide and protein level. In a last step DIAlignR for chromatogram alignment and quantification is carried out and a csv of peptide quantities, MSstats based protein statistics and several visualisations are exported.
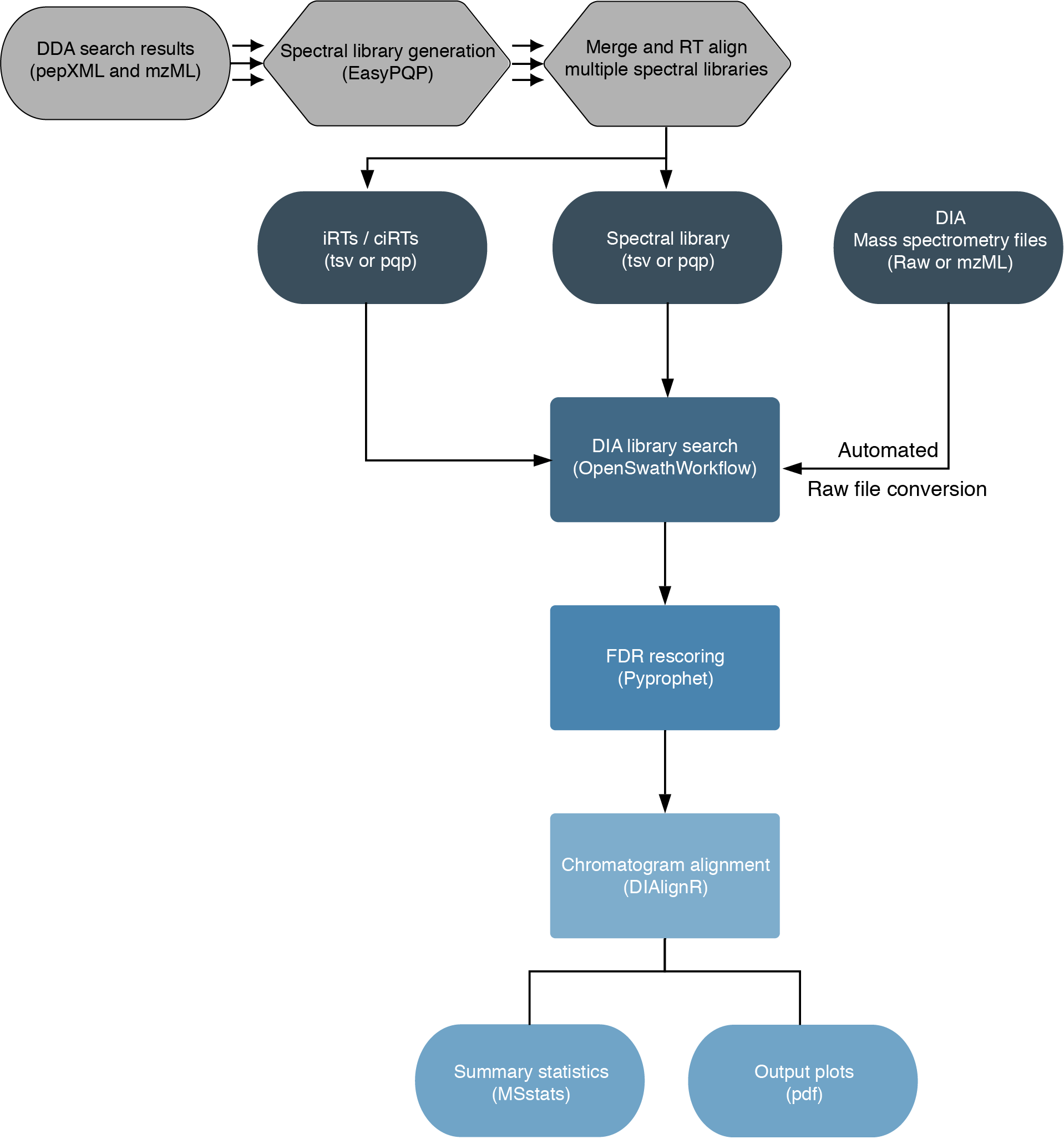 (This chart was created with the help of Lucidchart)
(This chart was created with the help of Lucidchart)
Quick Start
-
Install
nextflow -
Install any of
Docker,Singularity,Podman,ShifterorCharliecloudfor full pipeline reproducibility (please only useCondaas a last resort; see docs) -
Download the pipeline and test it on a minimal dataset with a single command:
nextflow run nf-core/diaproteomics -profile test,<docker/singularity/podman/shifter/charliecloud/conda/institute>Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment. -
Start running your own analysis!
nextflow run nf-core/diaproteomics -profile <docker/singularity/podman/conda/institute>--input 'sample_sheet.tsv'--input_spectral_library 'library_sheet.tsv'--irts 'irt_sheet.tsv'--mz_extraction_window 30--mz_extraction_window_unit ppm--rt_extraction_window 600--pyprophet_global_fdr_level 'protein'--pyprophet_protein_fdr 0.01Alternatively, create spectral libraries and iRTs:
nextflow run nf-core/diaproteomics -profile <docker/singularity/podman/conda/institute>--input 'sample_sheet.tsv'--generate_spectral_library--input_sheet_dda 'dda_sheet.tsv'--generate_pseudo_irts--merge_libraries--align_libraries
See usage docs for all of the available options when running the pipeline.
Pipeline Summary
By default, the pipeline currently performs the following:
- Optional spectral library generation from DDA input (‘EasyPQP’)
- DIA Targeted Extraction (‘OpenSwathWorkflow’)
- False discovery rate estimation (‘Pyprophet’)
- Chromatogram alignment (‘DIAlignR’)
- Statistical postprocessing (‘MSstats’)
Documentation
The nf-core/diaproteomics pipeline comes with documentation about the pipeline: usage and output.
Credits
nf-core/diaproteomics was originally written by Leon Bichmann.
We thank the following people for their extensive assistance in the development of this pipeline:
Shubham Gupta, George Rosenberger, Leon Kuchenbecker, Timo Sachsenberg, Oliver Alka, Julianus Pfeuffer and the nf-core team
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #diaproteomics channel (you can join with this invite).
Citations
If you apply DIAproteomics on your data please cite:
DIAproteomics: A multi-functional data analysis pipeline for data-independent-acquisition proteomics and peptidomics
Leon Bichmann, Shubham Gupta, George Rosenberger, Leon Kuchenbecker, Timo Sachsenberg, Oliver Alka, Julianus Pfeuffer, Oliver Kohlbacher & Hannes Rost.
bioRxiv: “https://www.biorxiv.org/content/10.1101/2020.12.08.415844v1”
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
In addition, references of tools and data used in this pipeline are as follows:
OpenSwathWorkflow
Röst H. et al, Nat Biotechnol. 2014 Mar;32(3):219-23. doi: 10.1038/nbt.2841.
PyProphet
Rosenberger G. et al, Nat Methods 2017 Sep;14(9):921-927. doi: 10.1038/nmeth.4398. Epub 2017 Aug 21.
DIAlignR
Gupta S. et al, Mol Cell Proteomics 2019 Apr;18(4):806-817. doi: 10.1074/mcp.TIR118.001132. Epub 2019 Jan 31.
MSstats
Choi M. et al, Bioinformatics 2014 Sep 1;30(17):2524-6. doi: 10.1093/bioinformatics/btu305. Epub 2014 May 2.