nf-core/diseasemodulediscovery
A pipeline for network-based disease module identification.
Introduction
nf-core/diseasemodulediscovery is a bioinformatics pipeline for network medicine hypothesis generation, designed for identifying active/disease modules. Developed and maintained by the RePo4EU consortium, it aims to characterize the molecular mechanisms of diseases by analyzing the local neighborhood of disease-associated genes or proteins (seeds) within the interactome. This approach can help identify potential drug targets for drug repurposing.
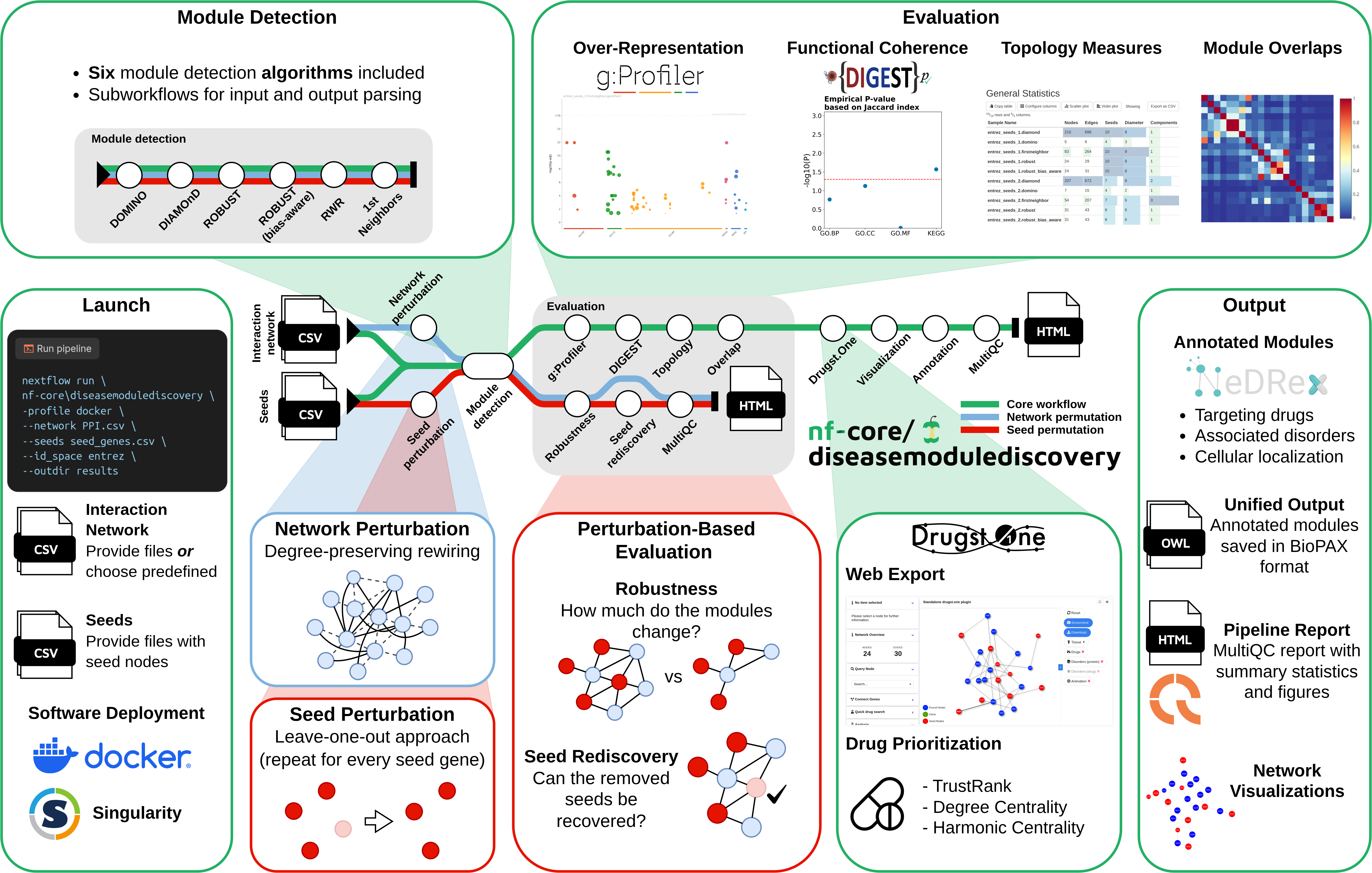
- Module inference (all enabled by default):
DOMINODIAMOnDROBUSTROBUST (bias-aware)1st Neighborsrandom walk with restart (RWR)
- Evaluation
- Over-representation analysis (
g:Profiler) - Functional coherence analysis (
DIGEST) - Network topology analysis (
graph-tool) - Overlaps between different disease modules
- Seed set perturbation-based evaluation (robustness and seed rediscovery, enabled by
--run_seed_perturbation) - Network perturbation-based evaluation (robustness, enabled by
--run_network_perturbation)
- Over-representation analysis (
- Export to the network medicine web visualization tool
Drugst.One - Drug prioritization using the API of
Drugst.One - Visualization of the module networks (
graph-tool,pyvis) - Annotation with biological data (targeting drugs, side effects, associated disorders, cellular localization) queried from
NeDRexDBand conversion toBioPAXformat. - Result and evaluation summary (
MultiQC)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Test your setup
nextflow run nf-core/diseasemodulediscovery \ -profile <docker/singularity>,test \ --outdir <OUTDIR>This will run the pipeline with a small test dataset. Results will be saved to the specified <OUTDIR>. Use -profile to set whether docker or singularity should be used for software deployment.
Running the pipeline
Now, you can run the pipeline with your own data using:
nextflow run nf-core/diseasemodulediscovery \ -profile <docker/singularity> \ --seeds <SEED_FILE> \ --network <NETWORK_FILE> \ --outdir <OUTDIR>This will run the pipeline based on the provided <SEED_FILE> and <NETWORK_FILE>. Results will be saved to the specified <OUTDIR>. Use -profile to set whether docker or singularity should be used for software deployment.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
OS specifics
The pipeline works best in combination with Linux. Furthermore, some Docker images in the pipeline are natively only available for amd64 but not the arm architecture.
Here are some tips to get the pipeline running with a different OS or architecture:
macOS
With macOS and Apple silicon, we had better experiences using the free version of orbstack instead of Docker Desktop for deploying the containers.
Windows
The most reliable solution is to work with the Windows Subsystem for Linux (WSL).
What if it keeps failing?
Most pipeline steps are not essential. If the pipeline keeps failing because of a specific process, you may be able to just skip that one.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/diseasemodulediscovery was originally written by the RePo4EU consortium.
We thank the following people for their extensive assistance in the development of this pipeline:
- Johannes Kersting (TUM)
- Lisa Spindler (TUM)
- Quirin Manz (TUM)
- Quim Aguirre (STALICLA)
- Chloé Bucheron (University Vienna)
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
If you want to include an additional module identification approach, please see this guide.
For further information or help, don’t hesitate to get in touch on the Slack #diseasemodulediscovery channel (you can join with this invite).
Citations
If you use nf-core/diseasemodulediscovery for your analysis, please cite the corresponding publication:
Johannes Kersting, Chloé Bucheron, Lisa M Spindler, Joaquim Aguirre-Plans, Quirin Manz, Tanja Pock, Mo Tan, Fernando M Delgado-Chaves, Cristian Nogales, Harald H H W Schmidt, Jörg Menche, Andreas Maier, Jan Baumbach, Emre Guney, Markus List, Inferring and evaluating network medicine-based disease modules with nextflow, Bioinformatics, Volume 42, Issue Supplement_1, July 2026, btag223, doi: 10.1093/bioinformatics/btag223
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.