nf-core/eager
A fully reproducible and state-of-the-art ancient DNA analysis pipeline
22.10.6.Learn more.Introduction
nf-core/eager is a scalable and reproducible bioinformatics best-practise processing pipeline for genomic NGS sequencing data, with a focus on ancient DNA (aDNA) data. It is ideal for the (palaeo)genomic analysis of humans, animals, plants, microbes and even microbiomes.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible. The pipeline pre-processes raw data from FASTQ inputs, or preprocessed BAM inputs. It can align reads and performs extensive general NGS and aDNA specific quality-control on the results. It comes with docker, singularity or conda containers making installation trivial and results highly reproducible.
Quick Start
-
Install
nextflow(>=20.07.1&&<=22.10.6) -
Install any of
Docker,Singularity,Podman,ShifterorCharliecloudfor full pipeline reproducibility (please only useCondaas a last resort; see docs) -
Download the pipeline and test it on a minimal dataset with a single command:
nextflow run nf-core/eager -profile test,<docker/singularity/podman/shifter/charliecloud/conda/institute>Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment. -
Start running your own analysis!
nextflow run nf-core/eager -profile <docker/singularity/podman/conda/institute> --input '*_R{1,2}.fastq.gz' --fasta '<your_reference>.fasta' -
Once your run has completed successfully, clean up the intermediate files.
nextflow clean -f -k
See usage docs for all of the available options when running the pipeline.
N.B. You can see an overview of the run in the MultiQC report located at ./results/MultiQC/multiqc_report.html
Modifications to the default pipeline are easily made using various options as described in the documentation.
Pipeline Summary
Default Steps
By default the pipeline currently performs the following:
- Create reference genome indices for mapping (
bwa,samtools, andpicard) - Sequencing quality control (
FastQC) - Sequencing adapter removal, paired-end data merging (
AdapterRemoval) - Read mapping to reference using (
bwa aln,bwa mem,CircularMapper, orbowtie2) - Post-mapping processing, statistics and conversion to bam (
samtools) - Ancient DNA C-to-T damage pattern visualisation (
DamageProfilerormapDamage) - PCR duplicate removal (
DeDuporMarkDuplicates) - Post-mapping statistics and BAM quality control (
Qualimap) - Library Complexity Estimation (
preseq) - Overall pipeline statistics summaries (
MultiQC)
Additional Steps
Additional functionality contained by the pipeline currently includes:
Input
- Automatic merging of complex sequencing setups (e.g. multiple lanes, sequencing configurations, library types)
Preprocessing
- Illumina two-coloured sequencer poly-G tail removal (
fastp) - Post-AdapterRemoval trimming of FASTQ files prior mapping (
fastp) - Automatic conversion of unmapped reads to FASTQ (
samtools) - Host DNA (mapped reads) stripping from input FASTQ files (for sensitive samples)
aDNA Damage manipulation
- Damage removal/clipping for UDG+/UDG-half treatment protocols (
BamUtil) - Damaged reads extraction and assessment (
PMDTools) - Nuclear DNA contamination estimation of human samples (
angsd)
Genotyping
- Creation of VCF genotyping files (
GATK UnifiedGenotyper,GATK HaplotypeCallerandFreeBayes) - Creation of EIGENSTRAT genotyping files (
pileupCaller) - Creation of Genotype Likelihood files (
angsd) - Consensus sequence FASTA creation (
VCF2Genome) - SNP Table generation (
MultiVCFAnalyzer)
Biological Information
- Mitochondrial to Nuclear read ratio calculation (
MtNucRatioCalculator) - Statistical sex determination of human individuals (
Sex.DetERRmine)
Metagenomic Screening
- Low-sequenced complexity filtering (
BBduk) - Taxonomic binner with alignment (
MALT) - Taxonomic binner without alignment (
Kraken2) - aDNA characteristic screening of taxonomically binned data from MALT (
MaltExtract)
Functionality Overview
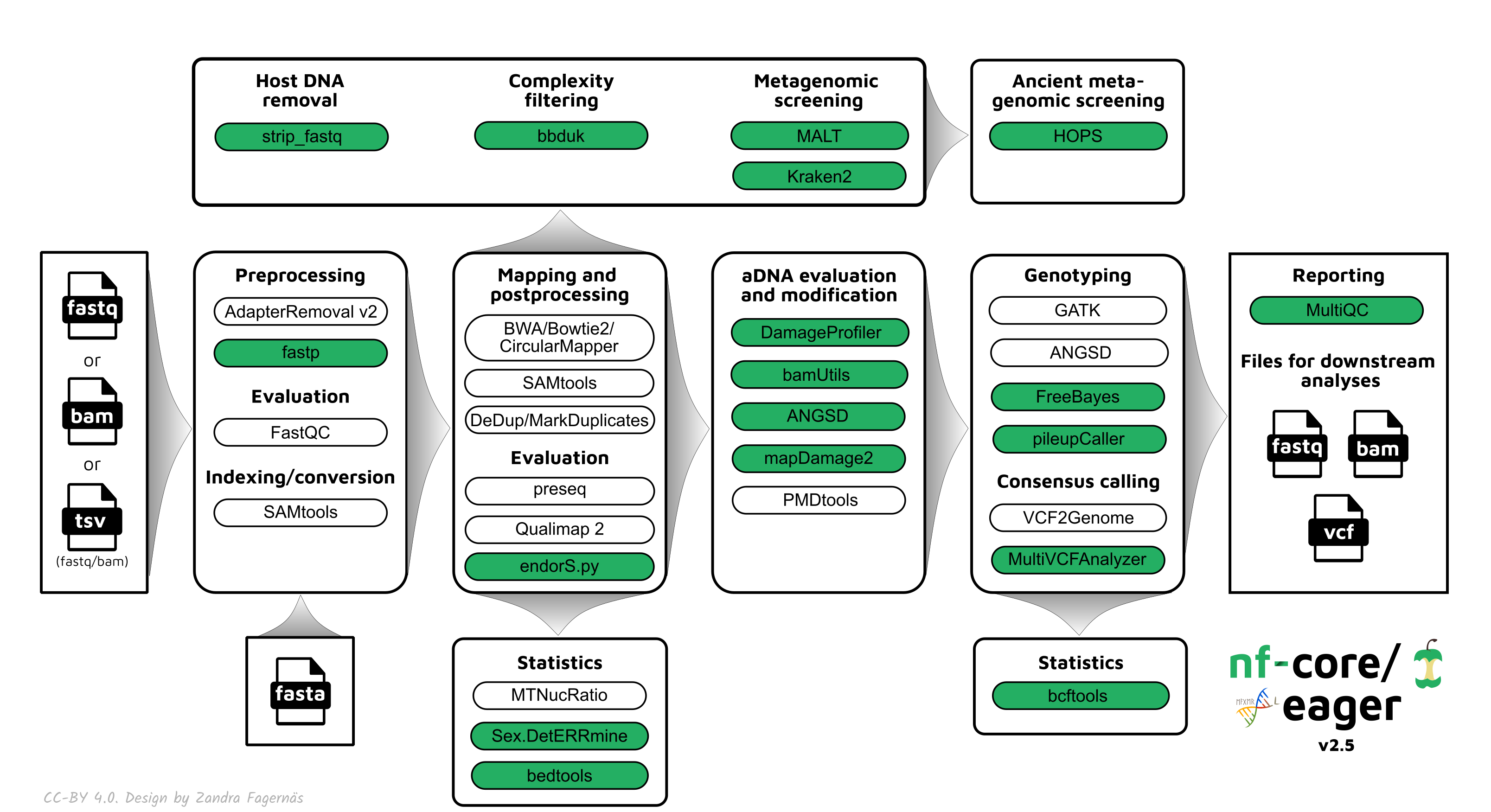
A graphical overview of suggested routes through the pipeline depending on context can be seen below.
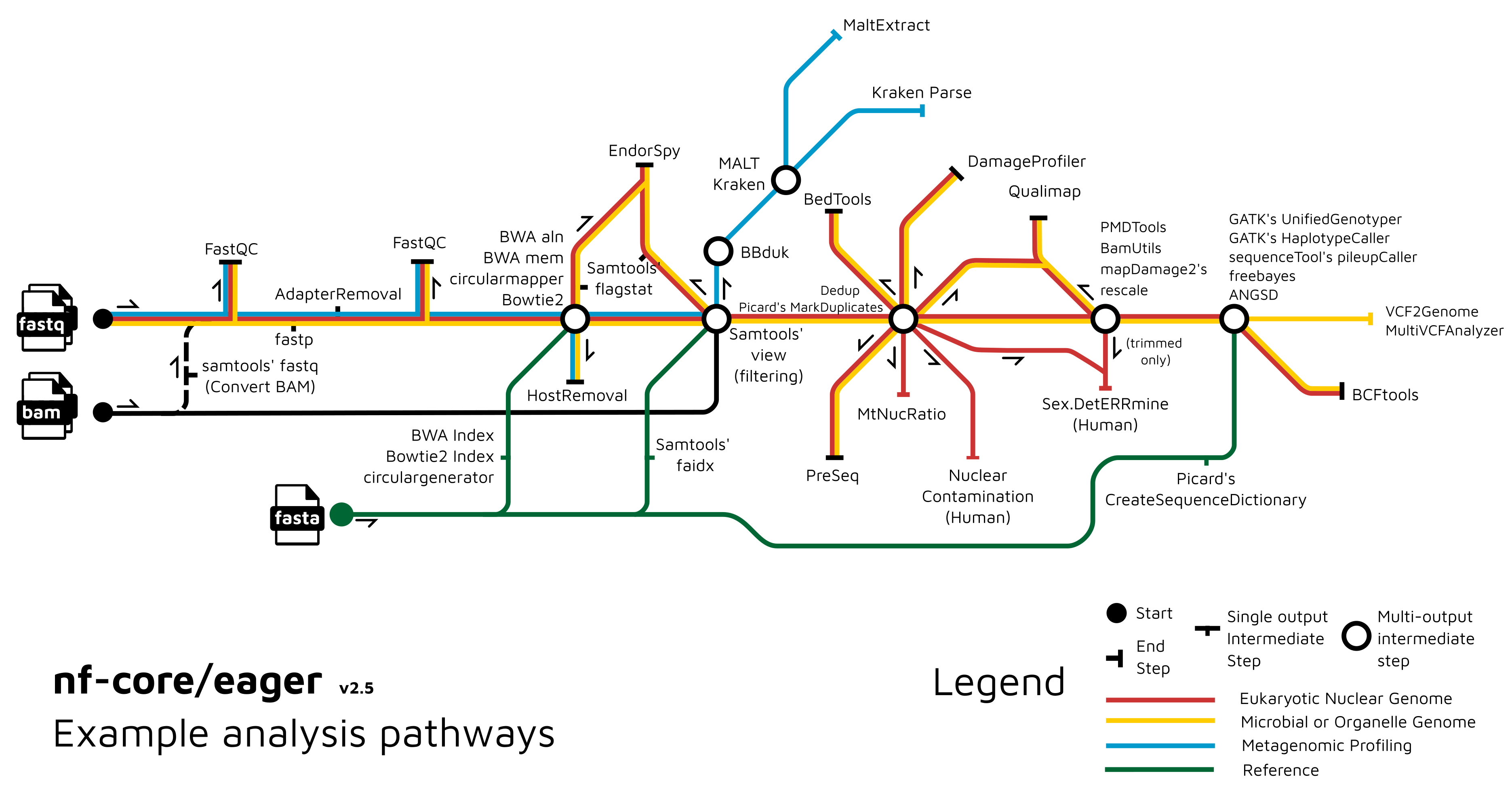
Documentation
The nf-core/eager pipeline comes with documentation about the pipeline: usage and output.
- Nextflow installation
- Pipeline configuration
- Running the pipeline
- This includes tutorials, FAQs, and troubleshooting instructions
- Output and how to interpret the results
Credits
This pipeline was mostly written by Alexander Peltzer (apeltzer) and James A. Fellows Yates, with contributions from Stephen Clayton, Thiseas C. Lamnidis, Maxime Borry, Zandra Fagernäs, Aida Andrades Valtueña and Maxime Garcia and the nf-core community.
We thank the following people for their extensive assistance in the development of this pipeline:
Authors (alphabetical)
- Aida Andrades Valtueña
- Alexander Peltzer
- James A. Fellows Yates
- Judith Neukamm
- Maxime Borry
- Maxime Garcia
- Stephen Clayton
- Thiseas C. Lamnidis
- Zandra Fagernäs
Additional Contributors (alphabetical)
Those who have provided conceptual guidance, suggestions, bug reports etc.
- Alex Hübner
- Alexandre Gilardet
- Arielle Munters
- Åshild Vågene
- Asmaa Ali
- Charles Plessy
- Elina Salmela
- Fabian Lehmann
- He Yu
- Hester van Schalkwyk
- Ido Bar
- Irina Velsko
- Işın Altınkaya
- Johan Nylander
- Jonas Niemann
- Katerine Eaton
- Kathrin Nägele
- Kevin Lord
- Laura Lacher
- Luc Venturini
- Mahesh Binzer-Panchal
- Marcel Keller
- Megan Michel
- Pierre Lindenbaum
- Pontus Skoglund
- Raphael Eisenhofer
- Roberta Davidson
- Rodrigo Barquera
- Selina Carlhoff
- Torsten Günter
If you’ve contributed and you’re missing in here, please let us know and we will add you in of course!
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #eager channel (you can join with this invite).
Citations
If you use nf-core/eager for your analysis, please cite the eager preprint as follows:
Fellows Yates JA, Lamnidis TC, Borry M, Valtueña Andrades A, Fagernäs Z, Clayton S, Garcia MU, Neukamm J, Peltzer A. 2021. Reproducible, portable, and efficient ancient genome reconstruction with nf-core/eager. PeerJ 9:e10947. DOI: 10.7717/peerj.10947.
You can cite the eager zenodo record for a specific version using the following doi: 10.5281/zenodo.3698082
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
In addition, references of tools and data used in this pipeline are as follows:
- EAGER v1, CircularMapper, DeDup* Peltzer, A., Jäger, G., Herbig, A., Seitz, A., Kniep, C., Krause, J., & Nieselt, K. (2016). EAGER: efficient ancient genome reconstruction. Genome Biology, 17(1), 1–14. https://doi.org/10.1186/s13059-016-0918-z. Download: https://github.com/apeltzer/EAGER-GUI and https://github.com/apeltzer/EAGER-CLI
- FastQC Download: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- AdapterRemoval v2 Schubert, M., Lindgreen, S., & Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Research Notes, 9, 88. https://doi.org/10.1186/s13104-016-1900-2. Download: https://github.com/MikkelSchubert/adapterremoval
- bwa Li, H., & Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics , 25(14), 1754–1760. https://doi.org/10.1093/bioinformatics/btp324. Download: http://bio-bwa.sourceforge.net/bwa.shtml
- SAMtools Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., … 1000 Genome Project Data Processing Subgroup. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics , 25(16), 2078–2079. https://doi.org/10.1093/bioinformatics/btp352. Download: http://www.htslib.org/
- DamageProfiler Neukamm, J., Peltzer, A., & Nieselt, K. (2020). DamageProfiler: Fast damage pattern calculation for ancient DNA. In Bioinformatics (btab190). https://doi.org/10.1093/bioinformatics/btab190. Download: https://github.com/Integrative-Transcriptomics/DamageProfiler
- QualiMap Okonechnikov, K., Conesa, A., & García-Alcalde, F. (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics , 32(2), 292–294. https://doi.org/10.1093/bioinformatics/btv566. Download: http://qualimap.bioinfo.cipf.es/
- preseq Daley, T., & Smith, A. D. (2013). Predicting the molecular complexity of sequencing libraries. Nature Methods, 10(4), 325–327. https://doi.org/10.1038/nmeth.2375. Download: http://smithlabresearch.org/software/preseq/
- PMDTools Skoglund, P., Northoff, B. H., Shunkov, M. V., Derevianko, A. P., Pääbo, S., Krause, J., & Jakobsson, M. (2014). Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proceedings of the National Academy of Sciences of the United States of America, 111(6), 2229–2234. https://doi.org/10.1073/pnas.1318934111. Download: https://github.com/pontussk/PMDtools
- MultiQC Ewels, P., Magnusson, M., Lundin, S., & Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics , 32(19), 3047–3048. https://doi.org/10.1093/bioinformatics/btw354. Download: https://multiqc.info/
- BamUtils Jun, G., Wing, M. K., Abecasis, G. R., & Kang, H. M. (2015). An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Research, 25(6), 918–925. https://doi.org/10.1101/gr.176552.114. Download: https://genome.sph.umich.edu/wiki/BamUtil
- FastP Chen, S., Zhou, Y., Chen, Y., & Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics , 34(17), i884–i890. https://doi.org/10.1093/bioinformatics/bty560. Download: https://github.com/OpenGene/fastp
- GATK 3.5 DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., … Daly, M. J. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 43(5), 491–498. https://doi.org/10.1038/ng.806.Download: https://console.cloud.google.com/storage/browser/gatk
- GATK 4.X - no citation available yet. Download: https://github.com/broadinstitute/gatk/releases
- VCF2Genome - Alexander Herbig and Alex Peltzer (unpublished). Download: https://github.com/apeltzer/VCF2Genome
- MultiVCFAnalyzer Bos, K.I. et al., 2014. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature, 514(7523), pp.494–497. Available at: http://dx.doi.org/10.1038/nature13591. Download: https://github.com/alexherbig/MultiVCFAnalyzer
- MTNucRatioCalculator Alex Peltzter (Unpublished). Download: https://github.com/apeltzer/MTNucRatioCalculator
- Sex.DetERRmine.py Lamnidis, T.C. et al., 2018. Ancient Fennoscandian genomes reveal origin and spread of Siberian ancestry in Europe. Nature communications, 9(1), p.5018. Available at: http://dx.doi.org/10.1038/s41467-018-07483-5. Download: https://github.com/TCLamnidis/Sex.DetERRmine.git
- ANGSD Korneliussen, T.S., Albrechtsen, A. & Nielsen, R., 2014. ANGSD: Analysis of Next Generation Sequencing Data. BMC bioinformatics, 15, p.356. Available at: http://dx.doi.org/10.1186/s12859-014-0356-4. Download: https://github.com/ANGSD/angsd
- bedtools Quinlan, A.R. & Hall, I.M., 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics , 26(6), pp.841–842. Available at: http://dx.doi.org/10.1093/bioinformatics/btq033. Download: https://github.com/arq5x/bedtools2/releases
- MALT. Download: https://software-ab.informatik.uni-tuebingen.de/download/malt/welcome.html
- Vågene, Å.J. et al., 2018. Salmonella enterica genomes from victims of a major sixteenth-century epidemic in Mexico. Nature ecology & evolution, 2(3), pp.520–528. Available at: http://dx.doi.org/10.1038/s41559-017-0446-6.
- Herbig, A. et al., 2016. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv, p.050559. Available at: http://biorxiv.org/content/early/2016/04/27/050559.
- MaltExtract Huebler, R. et al., 2019. HOPS: Automated detection and authentication of pathogen DNA in archaeological remains. bioRxiv, p.534198. Available at: https://www.biorxiv.org/content/10.1101/534198v1?rss=1. Download: https://github.com/rhuebler/MaltExtract
- Kraken2 Wood, D et al., 2019. Improved metagenomic analysis with Kraken 2. Genome Biology volume 20, Article number: 257. Available at: https://doi.org/10.1186/s13059-019-1891-0. Download: https://ccb.jhu.edu/software/kraken2/
- endorS.py Aida Andrades Valtueña (Unpublished). Download: https://github.com/aidaanva/endorS.py
- Bowtie2 Langmead, B. and Salzberg, S. L. 2012 Fast gapped-read alignment with Bowtie 2. Nature methods, 9(4), p. 357–359. doi: 10.1038/nmeth.1923.
- sequenceTools Stephan Schiffels (Unpublished). Download: https://github.com/stschiff/sequenceTools
- EigenstratDatabaseTools Thiseas C. Lamnidis (Unpublished). Download: https://github.com/TCLamnidis/EigenStratDatabaseTools.git
- mapDamage Jónsson, H., et al 2013. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics , 29(13), 1682–1684. https://doi.org/10.1093/bioinformatics/btt193
- BBduk Brian Bushnell (Unpublished). Download: https://sourceforge.net/projects/bbmap/
Data References
This repository uses test data from the following studies:
- Fellows Yates, J. A. et al. (2017) ‘Central European Woolly Mammoth Population Dynamics: Insights from Late Pleistocene Mitochondrial Genomes’, Scientific reports, 7(1), p. 17714. doi: 10.1038/s41598-017-17723-1.
- Gamba, C. et al. (2014) ‘Genome flux and stasis in a five millennium transect of European prehistory’, Nature communications, 5, p. 5257. doi: 10.1038/ncomms6257.
- Star, B. et al. (2017) ‘Ancient DNA reveals the Arctic origin of Viking Age cod from Haithabu, Germany’, Proceedings of the National Academy of Sciences of the United States of America, 114(34), pp. 9152–9157. doi: 10.1073/pnas.1710186114.
- de Barros Damgaard, P. et al. (2018). ‘137 ancient human genomes from across the Eurasian steppes.’, Nature, 557(7705), 369–374. doi: 10.1038/s41586-018-0094-2