nf-core/fetchngs
Pipeline to fetch metadata and raw FastQ files from public databases
Introduction
nf-core/fetchngs is a bioinformatics pipeline to fetch metadata and raw FastQ files from both public databases. At present, the pipeline supports SRA / ENA / DDBJ / GEO ids (see usage docs).
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
ids.csv:
SRR9984183SRR13191702ERR1160846ERR1109373DRR028935DRR026872Each line represents a database id. Please see next section for supported ids.
Now, you can run the pipeline using:
nextflow run nf-core/fetchngs \ -profile <docker/singularity/.../institute> \ --input ids.csv \ --outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Supported ids
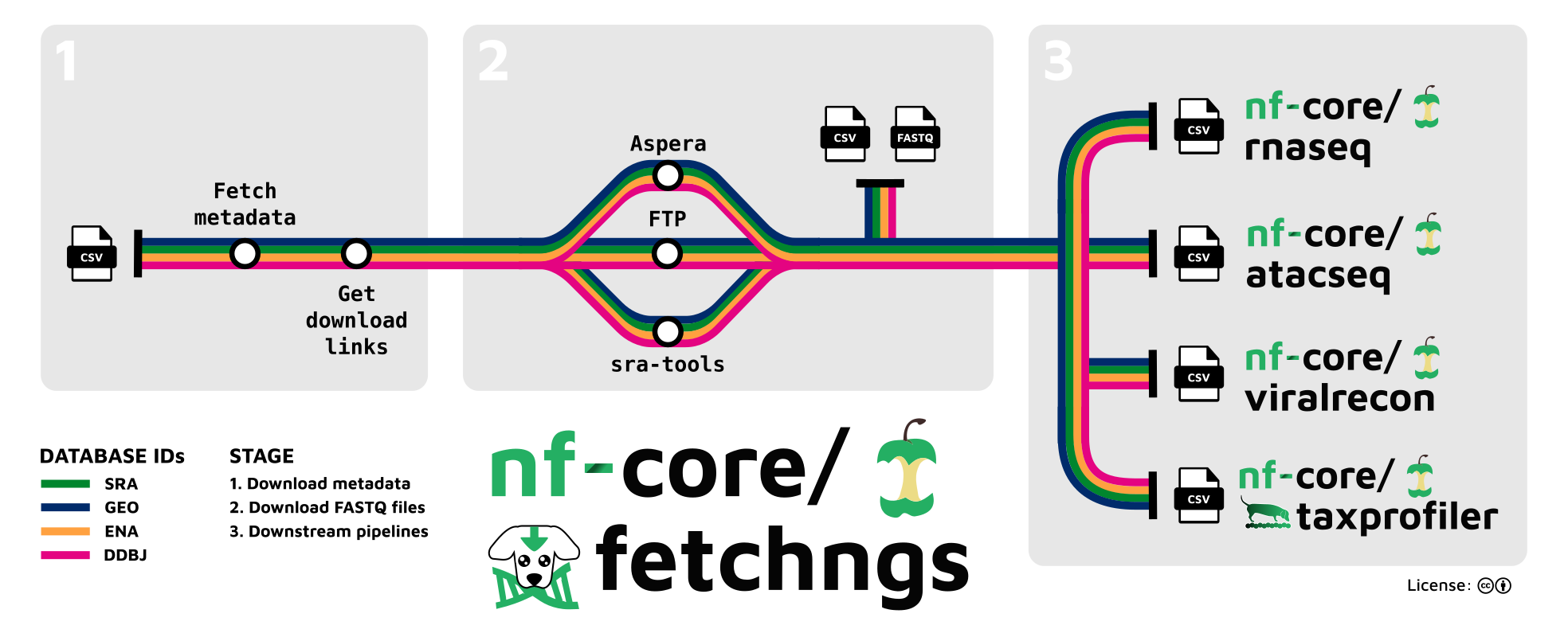
Via a single file of ids, provided one-per-line (see example input file) the pipeline performs the following steps:
SRA / ENA / DDBJ / GEO ids
- Resolve database ids back to appropriate experiment-level ids and to be compatible with the ENA API
- Fetch extensive id metadata via ENA API
- Download FastQ files:
- If direct download links are available from the ENA API:
- Fetch in parallel via
wgetand performmd5sumcheck (--download_method ftp; default). - Fetch in parallel via
aspera-cliand performmd5sumcheck. Use--download_method asperato force this behaviour.
- Fetch in parallel via
- Otherwise use
sra-toolsto download.srafiles and convert them to FastQ. Use--download_method sratoolsto force this behaviour.
- If direct download links are available from the ENA API:
- Collate id metadata and paths to FastQ files in a single samplesheet
Pipeline output
The columns in the output samplesheet can be tailored to be accepted out-of-the-box by selected nf-core pipelines (see usage docs), these currently include:
- nf-core/rnaseq
- nf-core/atacseq
- Ilumina processing mode of nf-core/viralrecon
- nf-core/taxprofiler
To see the the results of a test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/fetchngs was originally written by Harshil Patel (@drpatelh) from Seqera Labs, Spain and Jose Espinosa-Carrasco (@JoseEspinosa) from The Comparative Bioinformatics Group at The Centre for Genomic Regulation, Spain. Support for download of sequencing reads without FTP links via sra-tools was added by Moritz E. Beber (@Midnighter) from Unseen Bio ApS, Denmark.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #fetchngs channel (you can join with this invite).
Citations
If you use nf-core/fetchngs for your analysis, please cite it using the following doi: 10.5281/zenodo.5070524
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.