Usage
Pipeline parameters
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
Samplesheet input
You will need to create a samplesheet with information about the samples you would like to analyse before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with the 4 required columns: sample, fastq_1, fastq_2, and strandedness. If samples were sequenced on different platforms or at different centers, you can specify per-sample values via optional seq_platform and seq_center columns (used in BAM read group tags). For uniform values across all samples, the --seq_platform and --seq_center parameters are simpler. Please refer to the example below.
--input '[path to samplesheet file]'Multiple runs of the same sample
The sample identifiers have to be the same when you have re-sequenced the same sample more than once e.g. to increase sequencing depth. The pipeline will concatenate the raw reads before performing any downstream analysis. Below is an example for the same sample sequenced across 3 lanes.
sample,fastq_1,fastq_2,strandedness,seq_platformCONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,auto,ILLUMINACONTROL_REP1,AEG588A1_S1_L003_R1_001.fastq.gz,AEG588A1_S1_L003_R2_001.fastq.gz,auto,ILLUMINACONTROL_REP1,AEG588A1_S1_L004_R1_001.fastq.gz,AEG588A1_S1_L004_R2_001.fastq.gz,auto,ILLUMINALinting
By default, the pipeline will run fq lint on all input FASTQ files, both at the start of preprocessing and after each preprocessing step that manipulates FASTQ files. If errors are found, and error will be reported and the workflow will stop.
The extra_fqlint_args parameter can be manipulated to disable any validator from fq you wish. For example, we have found that checks on the names of paired reads are prone to failure, so that check is disabled by default (setting extra_fqlint_args to --disable-validator P001).
Strandedness Prediction
If you set the strandedness value to auto, the pipeline will sub-sample the input FastQ files to 1 million reads, use Salmon Quant to automatically infer the strandedness, and then propagate this information through the rest of the pipeline. This behavior is controlled by the --stranded_threshold and --unstranded_threshold parameters, which are set to 0.8 and 0.1 by default, respectively. This means:
- Forward stranded: At least 80% of the fragments are in the ‘forward’ orientation.
- Unstranded: The forward and reverse fractions differ by less than 10%.
- Undetermined: Samples that do not meet either criterion, possibly indicating issues such as genomic DNA contamination.
Note: These thresholds apply to both the strandedness inferred from Salmon outputs for input to the pipeline and how strandedness is inferred from RSeQC results using pipeline outputs.
Usage Examples
-
Forward Stranded Sample:
- Forward fraction: 0.85
- Reverse fraction: 0.15
- Classification: Forward stranded
-
Reverse Stranded Sample:
- Forward fraction: 0.1
- Reverse fraction: 0.9
- Classification: Reverse stranded
-
Unstranded Sample:
- Forward fraction: 0.45
- Reverse fraction: 0.55
- Classification: Unstranded
-
Undetermined Sample:
- Forward fraction: 0.6
- Reverse fraction: 0.4
- Classification: Undetermined
You can control the stringency of this behavior with --stranded_threshold and --unstranded_threshold.
Errors and Reporting
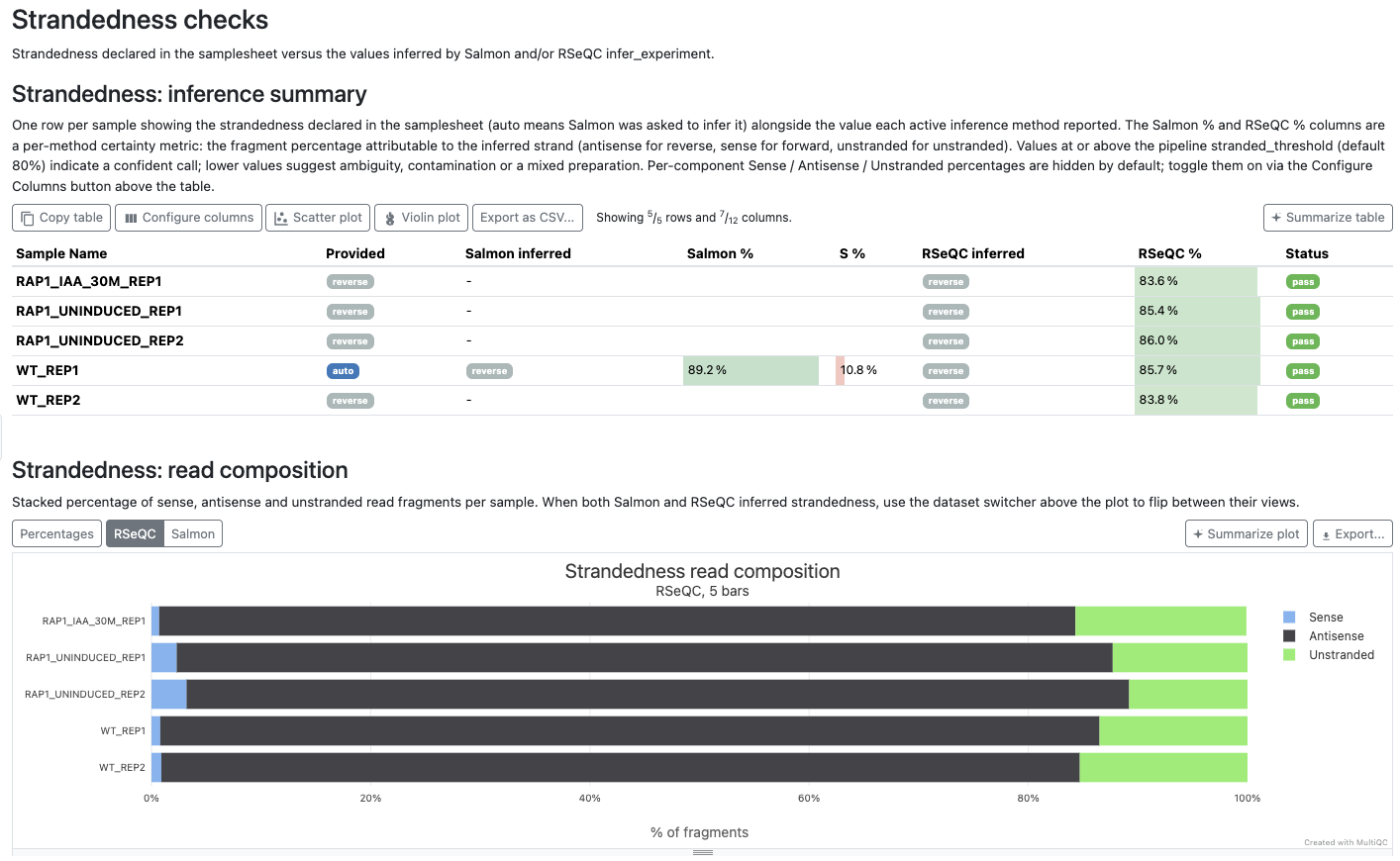
The results of strandedness inference are displayed in the MultiQC report under the top-level ‘Strandedness checks’ section, which contains two sub-reports:
- Strandedness: inference summary - one row per sample with the strandedness declared in the samplesheet, the values inferred by Salmon and RSeQC (where active), and a per-method certainty percentage (the fragment proportion attributable to the inferred strand). A pass/fail status is shown whenever RSeQC
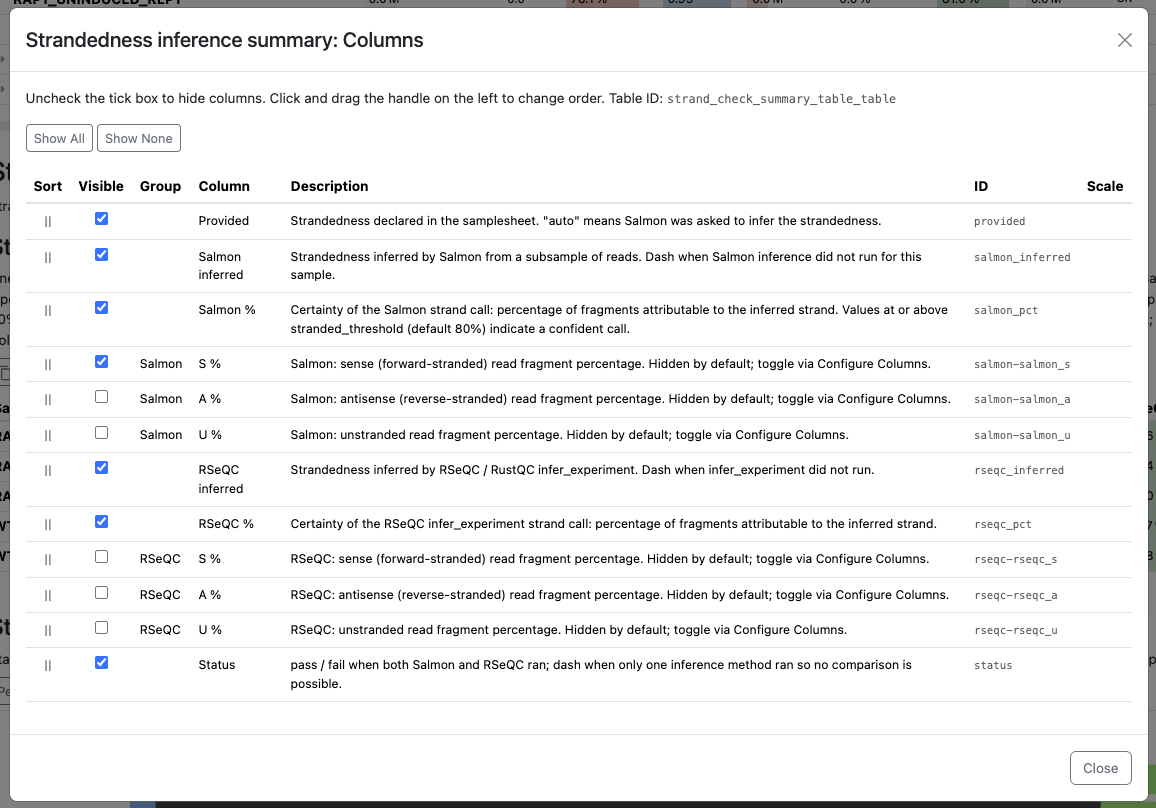
infer_experimentruns: RSeQC’s call is compared against Salmon’s auto-inference forautosamples and against the samplesheet value otherwise. The status column shows ‘-’ when RSeQC didn’t run and only the Salmon call is available. Per-component sense / antisense / unstranded percentages for each method are available as additional columns hidden by default - enable them via the Configure columns button above the table if you want to see the underlying composition. - Strandedness: read composition - stacked percentage bars of sense / antisense / unstranded fragments per sample. When both Salmon and RSeQC produced an inference, a dataset switcher above the plot lets you flip between the two methods’ views.
If RSeQC disagrees with the expected strandedness, or returns ‘undetermined’ (which can indicate gDNA contamination), the summary row is marked as a fail. When RSeQC isn’t active (e.g. --skip_rseqc, --skip_qc), Salmon’s auto-inference is still surfaced so the user always sees what the pipeline determined - just without a cross-check status.
The Configure columns dialog above the summary table lets you toggle the hidden per-component percentages on:
Be sure to check the strandedness section when reviewing the QC for your samples.
Full samplesheet
The pipeline will auto-detect whether a sample is single- or paired-end using the information provided in the samplesheet. The samplesheet can have as many columns as you desire, however, there is a strict requirement for the first 4 columns to match those defined in the table below.
A final samplesheet file consisting of both single- and paired-end data may look something like the one below. This is for 6 samples, where TREATMENT_REP3 has been sequenced twice.
sample,fastq_1,fastq_2,strandedness,seq_platformCONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,forward,ILLUMINACONTROL_REP2,AEG588A2_S2_L002_R1_001.fastq.gz,AEG588A2_S2_L002_R2_001.fastq.gz,forward,ILLUMINACONTROL_REP3,AEG588A3_S3_L002_R1_001.fastq.gz,AEG588A3_S3_L002_R2_001.fastq.gz,forward,ILLUMINATREATMENT_REP1,AEG588A4_S4_L003_R1_001.fastq.gz,,reverse,ILLUMINATREATMENT_REP2,AEG588A5_S5_L003_R1_001.fastq.gz,,reverse,ILLUMINATREATMENT_REP3,AEG588A6_S6_L003_R1_001.fastq.gz,,reverse,ILLUMINATREATMENT_REP3,AEG588A6_S6_L004_R1_001.fastq.gz,,reverse,ILLUMINA| Column | Description |
|---|---|
sample |
Custom sample name. This entry will be identical for multiple sequencing libraries/runs from the same sample. Spaces in sample names are automatically converted to underscores (_). |
fastq_1 |
Full path to FastQ file for Illumina short reads 1. File has to be gzipped and have the extension “.fastq.gz” or “.fq.gz”. |
fastq_2 |
Full path to FastQ file for Illumina short reads 2. File has to be gzipped and have the extension “.fastq.gz” or “.fq.gz”. |
strandedness |
Sample strand-specificity. Must be one of unstranded, forward, reverse or auto. |
seq_platform |
Optional. Sequencing platform for BAM read group PL tag (e.g., ILLUMINA). Use this column when samples were sequenced on different platforms. For a single platform across all samples, prefer --seq_platform instead. Per-sample values override the global parameter. |
seq_center |
Optional. Sequencing center for BAM read group CN tag. Use this column when samples come from different centers. For a single center across all samples, prefer --seq_center instead. Per-sample values override the global parameter. |
genome_bam |
Optional. Full path to genome-aligned BAM file. Typically from previous pipeline runs (see output documentation or STAR/RSEM). |
transcriptome_bam |
Optional. Full path to transcriptome-aligned BAM file. Typically from previous pipeline runs (see output documentation or STAR/RSEM). |
percent_mapped |
Optional. Percentage of reads that mapped during alignment (0-100). Useful for quality assessment and filtering. |
An example samplesheet has been provided with the pipeline.
NB: The
groupandreplicatecolumns were replaced with a singlesamplecolumn as of v3.1 of the pipeline. Thesamplecolumn is essentially a concatenation of thegroupandreplicatecolumns, however it now also offers more flexibility in instances where replicate information is not required e.g. when sequencing clinical samples. If all values ofsamplehave the same number of underscores, fields defined by these underscore-separated names may be used in the PCA plots produced by the pipeline, to regain the ability to represent different groupings.
BAM input for reprocessing workflow
The pipeline supports a two-step workflow for efficient reprocessing without expensive alignment steps. This feature is designed specifically for re-running with BAM files generated by previous runs of this same pipeline.
Step 1: Initial run with BAM generation
Run the pipeline normally, adding --save_align_intermeds to publish BAM files and generate a reusable samplesheet:
nextflow run nf-core/rnaseq \ --input samplesheet.csv \ --save_align_intermeds \ --outdir results_initial \ -profile dockerThis creates samplesheets/samplesheet_with_bams.csv containing paths to the generated BAM files.
Step 2: Reprocessing run with BAM input
Use the auto-generated samplesheet to reprocess data, skipping alignment:
nextflow run nf-core/rnaseq \ --input samplesheets/samplesheet_with_bams.csv \ --skip_alignment \ --outdir results_reprocessed \ -profile dockerThe --skip_alignment flag tells the pipeline to skip alignment, and in this situation it will use any provided BAM files instead of performing alignment, putting them through post-processing and quantification only.
Example of generated samplesheet
The samplesheet_with_bams.csv will look like:
sample,fastq_1,fastq_2,strandedness,seq_platform,seq_center,genome_bam,percent_mapped,transcriptome_bamSAMPLE1,/path/sample1_R1.fastq.gz,/path/sample1_R2.fastq.gz,forward,ILLUMINA,,results/star_salmon/SAMPLE1.markdup.sorted.bam,85.2,results/star_salmon/SAMPLE1.Aligned.toTranscriptome.out.bamSAMPLE2,/path/sample2_R1.fastq.gz,,reverse,ILLUMINA,,results/star_salmon/SAMPLE2.sorted.bam,92.1,results/star_salmon/SAMPLE2.Aligned.toTranscriptome.out.bamImportant limitations
⚠️ Warning: This feature is designed specifically for BAM files generated by this pipeline. Using arbitrary BAM files from other sources is not officially supported and will likely only work via the two-step workflow described above. Users attempting to use other BAMs do so at their own risk.
⚠️ Warning: You cannot mix quantifier types between BAM generation and reprocessing runs. BAM files generated with
--aligner star_salmonmust be reprocessed with--aligner star_salmon. Similarly, BAM files from--aligner star_rsemmust be reprocessed with--aligner star_rsem. Mixing quantifier types will likely produce incorrect results due to incompatible alignment parameters.
Key technical details:
- BAM files are only used when
--skip_alignmentis specified - The pipeline automatically indexes provided BAM files
- You can provide just
genome_bam, justtranscriptome_bam, or both - Mixed samplesheets are supported, but samples with BAM files require
--skip_alignment - Without
--skip_alignment, the pipeline will perform alignment even if BAM files are provided - For BAM file locations from pipeline outputs, see the output documentation
This workflow is ideal for tweaking downstream processing steps (quantification methods, QC parameters, differential expression analysis) without repeating time-consuming alignment.
FASTQ sampling
If you would like to reduce the number of reads used in the analysis, for example to test pipeline operation with limited resource usage, you can make use of the FASTP option for trimming (see below). FASTP has an option to take the first n reads of input FASTQ file(s), so this can be used to reduce the reads passed to subsequent steps. For example, to pass only the first 10,000 reads for trimming you would set input paramters like:
--trimmer fastp --extra_fastp_args '--reads_to_process 10000'Adapter trimming options
Trim Galore! is a wrapper tool around Cutadapt and FastQC to peform quality and adapter trimming on FastQ files. Trim Galore! will automatically detect and trim the appropriate adapter sequence. It is the default trimming tool used by this pipeline, however you can use fastp instead by specifying the --trimmer fastp parameter. fastp is a tool designed to provide fast, all-in-one preprocessing for FastQ files. It has been developed in C++ with multithreading support to achieve higher performance. You can specify additional options for Trim Galore! and fastp via the --extra_trimgalore_args and --extra_fastp_args parameters, respectively.
NB: TrimGalore! will only run using multiple cores if you are able to use more than > 5 and > 6 CPUs for single- and paired-end data, respectively. The total cores available to TrimGalore! will also be capped at 4 (7 and 8 CPUs in total for single- and paired-end data, respectively) because there is no longer a run-time benefit. See release notes and discussion whilst adding this logic to the nf-core/atacseq pipeline.
rRNA removal options
Ribosomal RNA (rRNA) removal can be enabled with the --remove_ribo_rna parameter. The pipeline supports three different tools for rRNA removal, selectable via the --ribo_removal_tool parameter.
For tools that use a reference database (SortMeRNA and Bowtie2), although rRNA is the primary target, the reference database can include additional abundant contaminant sequences you wish to remove, such as tRNAs or other non-coding RNAs. Simply add the paths to your custom FASTA files in the manifest file.
SortMeRNA (default)
SortMeRNA uses k-mer matching against rRNA databases to identify and filter rRNA reads. This is the default option and requires an rRNA database manifest file.
nextflow run nf-core/rnaseq --remove_ribo_rna --ribo_removal_tool sortmerna ...By default, the pipeline uses smr_v4.3_fast_db, a SILVA 138 and Rfam build clustered at 85/90 % (SILVA) and 90 % (Rfam). It covers archaeal, bacterial and eukaryotic 16S/18S/23S/28S rRNAs plus 5S/5.8S. The URL is listed in assets/rrna-db-defaults.txt, which is the default value of the --ribo_database_manifest parameter. SILVA 138+ is distributed under CC-BY 4.0, which permits commercial use with attribution.
SortMeRNA publishes four pre-built databases on its v4.3.7 release. They are the same underlying SILVA 138 + Rfam sequences progressively clustered: coarser clustering means a smaller reference, a smaller index and a faster run, at the cost of some sensitivity. The pipeline decompresses the gzipped FASTA before use.
| Variant | Clustering | Asset (gz) | Use when |
|---|---|---|---|
smr_v4.3_fast_db |
SILVA 85/90 %, Rfam 90 % | ~17 MB | Pipeline default. Routine bulk RNA-seq with good-quality input |
smr_v4.3_default_db |
SILVA 90/95 %, Rfam 95 % | ~36 MB | SortMeRNA upstream’s balanced recommendation; moderate extra index footprint |
smr_v4.3_sensitive_db |
SILVA 97 %, Rfam 97 % | ~93 MB | High-rRNA samples (e.g. total RNA without rRNA depletion), rare-lineage organisms |
smr_v4.3_sensitive_db_rfam_seeds |
SILVA 97 % + Rfam seeds | ~90 MB | Maximum sensitivity, including the full Rfam seed set for non-rRNA ncRNA families |
Picking a variant: fast keeps the index footprint close to what earlier pipeline versions used (~1-2 GB); sensitive and sensitive_rfam_seeds roughly triple that. Index size is a one-off cost per work directory (results are cached), but per-sample SortMeRNA memory and runtime also scale with the reference.
To switch variant or use your own references, write a manifest file listing one FASTA URL or local path per line and pass it via --ribo_database_manifest my_manifest.txt. For example, a one-line manifest selecting the default variant would contain https://github.com/sortmerna/sortmerna/releases/download/v4.3.7/smr_v4.3_default_db.fasta.gz.
Both plain .fasta and .fasta.gz entries are accepted; gzipped files are decompressed automatically before being handed to SortMeRNA.
Bowtie2
Bowtie2 performs alignment-based filtering against rRNA reference sequences. Reads that align to the rRNA references are filtered out, and unaligned reads are kept for downstream analysis. This option also requires an rRNA database manifest file specified via --ribo_database_manifest.
nextflow run nf-core/rnaseq --remove_ribo_rna --ribo_removal_tool bowtie2 ...RiboDetector
RiboDetector has known issues with ONNX multiprocessing that can cause hangs in containerized environments (Docker, Singularity). This makes it unreliable for production use in Nextflow pipelines. We recommend using SortMeRNA or Bowtie2 for rRNA removal until these issues are resolved upstream. See hzi-bifo/RiboDetector#61 for details.
RiboDetector uses machine learning to identify rRNA reads without requiring a reference database. This makes it particularly useful when working with organisms that lack well-characterized rRNA sequences, or when you want to avoid database licensing requirements.
nextflow run nf-core/rnaseq --remove_ribo_rna --ribo_removal_tool ribodetector ...RiboDetector automatically determines read length from your data and uses its pre-trained neural network model to classify reads.
GPU acceleration for RiboDetector
RiboDetector supports GPU acceleration via --use_gpu_ribodetector. When enabled, the pipeline uses a CUDA-enabled container with PyTorch GPU support and switches to the GPU binary automatically.
nextflow run nf-core/rnaseq --remove_ribo_rna --ribo_removal_tool ribodetector --use_gpu_ribodetector ...Requirements:
- One or more NVIDIA GPUs (with appropriate drivers installed)
- A container runtime (Docker, Singularity, or Apptainer); Conda is also supported
- x86_64 architecture (ARM GPU is not currently supported)
Container GPU flags (--gpus all for Docker, --nv for Singularity/Apptainer) are automatically applied and can be overridden via --gpu_container_options.
Alignment options
By default, the pipeline uses STAR (i.e. --aligner star_salmon) to map the raw FastQ reads to the reference genome, project the alignments onto the transcriptome and to perform the downstream BAM-level quantification with Salmon. STAR is fast but requires a lot of memory to run, typically around 38GB for the Human GRCh37 reference genome. Both --aligner star_salmon and --aligner star_rsem use STAR for alignment, so you should use the HISAT2 aligner (i.e. --aligner hisat2) if you have memory limitations.
Selecting star_rsem automatically applies the same STAR settings as rsem-calculate-expression with the --star option. These are based on the ENCODE3 STAR settings, and are as follows:
View STAR parameters
--outSAMunmapped Within--outFilterType BySJout--outFilterMultimapNmax 20--outFilterMismatchNmax 999--outFilterMismatchNoverLmax 0.04--alignIntronMin 20--alignIntronMax 1000000--alignMatesGapMax 1000000--alignSJoverhangMin 8--alignSJDBoverhangMin 1--sjdbScore 1NOTE: The pipeline parameter --extra_star_align_args cannot be used with aligner option star_rsem. It is possible to set, via custom tool arguments, custom settings to the process STAR_ALIGN via ext.args. However, we discourage this and consider adjusting the STAR aligner while using RSEM to be an unsupported option in this pipeline. If you need to pass in custom star alignment options, we recommend using the aligner option star_salmon. After this warning, if you still wish to set custom STAR settings and use RSEM for quantification, then please reference this configuration file.
You also have the option to pseudoalign and quantify your data directly with Salmon or Kallisto by specifying salmon or kallisto to the --pseudo_aligner parameter. The selected pseudoaligner will then be run in addition to the standard alignment workflow defined by --aligner, mainly because it allows you to obtain QC metrics with respect to the genomic alignments. However, you can provide the --skip_alignment parameter if you would like to run Salmon or Kallisto in isolation. By default, the pipeline will use the genome fasta and gtf file to generate the transcripts fasta file, and then to build the Salmon index. You can override these parameters using the --transcript_fasta and --salmon_index parameters, respectively.
The library preparation protocol (library type) used by Salmon quantification is inferred by the pipeline based on the information provided in the samplesheet, however, you can override it using the --salmon_quant_libtype parameter. You can find the available options in the Salmon documentation. Similarly, strandedness is taken from the sample sheet or calculated automatically, and passed to Kallisto on a per-library basis, but you can apply a global override by setting the Kallisto strandedness parameters in --extra_kallisto_quant_args like --extra_kallisto_quant_args '--fr-stranded' see the Kallisto documentation.
When running Salmon in mapping-based mode via --pseudo_aligner salmon, supplying a genome fasta via --fasta and not supplying a Salmon index, the entire genome of the organism is used by default for the decoy-aware transcriptome when creating the indices, as is recommended (see second bulleted option in Salmon documentation). If you do not supply a FASTA file or an index, Salmon will index without those decoys, using only transcript sequences in the index. This second option is not usually recommended, but may be useful in limited circumstances. Note that Kallisto does not index with genomic sequences.
Two additional parameters --extra_star_align_args and --extra_salmon_quant_args were added in v3.10 of the pipeline that allow you to append any custom parameters to the STAR align and Salmon quant commands, respectively. Note, the --seqBias and --gcBias are not provided to Salmon quant by default so you can provide these via --extra_salmon_quant_args '--seqBias --gcBias' if required. You can now also supply additional arguments to Kallisto via --extra_kallisto_quant_args.
You can use --skip_alignment --skip_pseudo_alignment if you only want to run the pre-processing QC steps in the pipeline like FastQ, trimming etc. This will skip alignment, pseudoalignment and any post-alignment processing steps.
Note that --skip_alignment and --skip_pseudo_alignment prevent both the execution of alignment/pseudoalignment steps and the building of their corresponding indices. For example, using --skip_alignment with --aligner star_salmon will skip both STAR alignment and index building.
Sentieon acceleration for STAR
The STAR aligner can be accelerated through its Sentieon implemention using the parameter --use_sentieon_star.
Sentieon is a commercial solution to process genomics data, requiring a paid license. Sentieon’s tooling contains an accelerated version of the STAR aligner, which nf-core/rnaseq supports. In order to use those functions, the user will need to supply a license for Sentieon.
Sentieon supply license in the form of a string-value (a url) or a file. It should be base64-encoded and stored in a nextflow secret named SENTIEON_LICENSE_BASE64. If a license string (url) is supplied, then the nextflow secret should be set like this:
nextflow secrets set SENTIEON_LICENSE_BASE64 $(echo -n <sentieon_license_string> | base64 -w 0)<sentieon_license_string> is formatted as IP:Port for example: 12.12.12.12:8990
If a license file is supplied, then the nextflow secret should be set like this:
nextflow secrets set SENTIEON_LICENSE_BASE64 \$(cat <sentieon_license_file.lic> | base64 -w 0)If you’re looking for documentation on how the nf-core Sentieon GitHub Actions and Sentieon License Server are set up: Here be dragons.
For detailed instructions on how to test the modules and subworkflows separately, see here.
Parabricks GPU acceleration for STAR
The STAR aligner can also be GPU-accelerated using NVIDIA Parabricks via the --use_parabricks_star parameter. Parabricks runs STAR alignment on NVIDIA GPUs, significantly reducing wall-clock time for large datasets.
nextflow run nf-core/rnaseq \ --input samplesheet.csv \ --outdir results \ --fasta genome.fa \ --gtf annotation.gtf \ --use_parabricks_star \ -profile dockerContainer GPU flags (--gpus all for Docker, --nv for Singularity/Apptainer) are automatically applied to GPU tasks based on the container engine. These flags are scoped to GPU tasks only, so non-GPU steps will run normally on CPU-only nodes in mixed clusters.
The container GPU flags can be overridden via --gpu_container_options (e.g. --gpu_container_options '--gpus 1').
Requirements
- One or more NVIDIA GPUs (with appropriate drivers installed)
- Docker or Singularity (Conda/Mamba is not supported for this module)
- The Parabricks container (
nvcr.io/nvidia/clara/clara-parabricks:4.6.0-1) will be pulled automatically
Behaviour differences
When using Parabricks, the pipeline automatically handles mark duplicates during the alignment step (via pbrun rna_fq2bam), so the separate Picard MarkDuplicates step is skipped.
Known differences from native STAR
Parabricks rna_fq2bam is based on STAR 2.7.2a. The following native STAR flags have no pbrun equivalent and are therefore not applied:
--outFilterType BySJout— pbrun uses its own splice junction filtering defaults--sjdbScore 1— affects junction scoring priority--quantTranscriptomeBan Singleend— Salmon handles mixed single/paired-end transcriptome records gracefully--runRNGseed 0— pbrun uses deterministic primary alignment selection
These differences are unlikely to materially affect downstream quantification results, but users should be aware of them for reproducibility purposes. All other STAR parameters (multi-mapping limits, intron sizes, mate gap, splice junction overhangs, etc.) have pbrun equivalents and are applied consistently.
RustQC: accelerated post-alignment QC (experimental)
RustQC support is experimental. We recommend trialling it alongside the default QC path on a pilot subset of your data before switching over for production runs. Results are designed to match the upstream tools, but minor numerical differences exist in some outputs (see below).
RustQC is a high-performance tool that replaces multiple post-alignment QC steps with a single pass over the BAM file. It produces output files compatible with the same MultiQC modules, so the report content is equivalent.
RustQC replaces the following tools: dupRadar, featureCounts (biotype QC), RSeQC (bam_stat, infer_experiment, read_distribution, read_duplication, junction_annotation, junction_saturation, inner_distance, TIN), Preseq, Qualimap, and SAMtools stats/flagstat/idxstats.
To enable RustQC, use the --use_rustqc flag. The individual tools listed above are automatically skipped:
nextflow run nf-core/rnaseq \ --input samplesheet.csv \ --outdir results \ --fasta genome.fa \ --gtf annotation.gtf \ --use_rustqc \ -profile dockerRustQC outputs are published under <ALIGNER>/rustqc/ with subdirectories matching the tools they replace (e.g. rustqc/rseqc/bam_stat/, rustqc/qualimap/). See the output documentation for the full list of files.
Preseq and TIN
The nf-core/rnaseq pipeline does not normally run Preseq and tin.py by default, as these QC steps are very slow and error prone. They are included in RustQC by default however, so these results will be produced when using --use_rustqc.
The upstream tools can be enabled with the following options:
--skip_preseq false \--rseqc_modules bam_stat,inner_distance,infer_experiment,junction_annotation,junction_saturation,read_distribution,read_duplication,tinValidation status
RustQC has been validated against the default QC tools on the nf-core/rnaseq test dataset (5 yeast samples, mixed PE/SE). The majority of outputs are byte-for-byte identical. Where differences exist (e.g. floating-point precision in curve fitting, randomized subsampling in TIN, and cosmetic formatting), they are minor and do not affect biological interpretation or MultiQC report content.
Quantification options
The current options align with STAR and quantify using either Salmon (--aligner star_salmon) / RSEM (--aligner star_rsem). You also have the option to pseudoalign and quantify your data with Salmon or Kallisto by providing the --pseudo_aligner salmon or --pseudo_aligner kallisto parameter, respectively.
Since v3.0 of the pipeline, featureCounts is no longer used to perform gene/transcript quantification, however it is still used to generate QC metrics based on biotype information available within GFF/GTF genome annotation files. This decision was made primarily because of the limitations of featureCounts to appropriately quantify gene expression data. Please see Zhao et al., 2015 and Soneson et al., 2015.
For similar reasons, quantification will not be performed if using --aligner hisat2 due to the lack of an appropriate option to calculate accurate expression estimates from HISAT2 derived genomic alignments - this may change in future releases (see #822). HISAT2 has been made available for those who have a preference for the alignment, QC and other types of downstream analysis compatible with it’s output.
Per-sample quantification (--skip_quantification_merge)
By default, the pipeline merges quantification results across all samples, producing cross-sample count matrices, SummarizedExperiment objects, and a single merged MultiQC report. This works well for typical experiments, but can become a bottleneck for very large cohorts (hundreds to thousands of samples) where:
- Memory: aggregation steps (tximport
.collect(), SummarizedExperiment, RSEM merge counts, merged MultiQC) load all samples into a single process, which can exceed available memory - Organization: run-centric output directories (
fastqc/,salmon/,multiqc/) mix files from all samples, making it harder to locate, deliver, or archive results per sample
The --skip_quantification_merge parameter switches the pipeline to sample-centric operation. Each sample is processed independently and all its outputs are organized under <outdir>/<sample_id>/:
results/ SAMPLE_A/ fastqc/ trimgalore/ salmon/ # per-sample quant + tximport TSVs multiqc/star_salmon/ # per-sample MultiQC report SAMPLE_B/ ... genome/ # shared reference files pipeline_info/ # run metadataWhen enabled:
- tximport runs per-sample, producing individual gene-level and transcript-level count/TPM TSVs for each sample
- The
*gene_counts_length_scaled.tsvfile is not published, because per-samplelengthScaledTPMvalues are not comparable across samples (#1822). To obtain it, re-run tximport across allquant.sffiles in a single call - SummarizedExperiment and RSEM merge counts are skipped entirely
- DESeq2 QC is skipped (requires multiple samples)
- MultiQC generates one report per sample rather than one merged report. Per-sample reports carry a manifest-only software versions section (pipeline name and Nextflow version); the full collated tool versions YAML is still published unchanged to
pipeline_info/
This mode works with any aligner or pseudo-aligner. For the fastest possible per-sample quantification, combine it with pseudo-alignment only:
nextflow run nf-core/rnaseq \ --input samplesheet.csv \ --pseudo_aligner salmon \ --skip_alignment \ --skip_quantification_merge \ --outdir resultsA convenience profile rapid_quant bundles these options together with additional skips for alignment-dependent QC steps:
nextflow run nf-core/rnaseq \ --input samplesheet.csv \ -profile rapid_quant,docker \ --outdir resultsPer-sample tximport TSVs can be combined downstream using standard R or Python tooling if cross-sample matrices are needed later. This gives you flexibility to run the pipeline incrementally as new samples arrive without reprocessing the entire cohort. Note that lengthScaledTPM cannot simply be concatenated and must be recomputed by passing all quant.sf files to a single tximport call.
Unique Molecular Identifiers (UMI)
The pipeline supports Unique Molecular Identifiers to increase the accuracy of the quantification. UMIs are short sequences used to uniquely tag each molecule in a sample library and facilitate the accurate identification of read duplicates. They must be added during library preparation and prior to sequencing, therefore require appropriate arrangements with your sequencing provider.
To take UMIs into consideration during a workflow run, specify the --with_umi parameter. The pipeline currently supports UMIs, which are embedded within a read’s sequence and UMIs, whose sequence is given inside the read’s name. Please consult your kit’s manual and/or contact your sequencing provider regarding the exact specification.
The --umitools_grouping_method parameter affects how similar, but non-identical UMIs are treated. directional, the default setting, is most accurate, but computationally very demanding. Consider percentile or unique if processing many samples.
Examples:
| UMI type | Source | Pipeline parameters |
|---|---|---|
| In read name | Illumina BCL convert >3.7.5 | --with_umi --skip_umi_extract --umitools_umi_separator ":" |
| In sequence | Lexogen QuantSeq® 3’ mRNA-Seq V2 FWD + UMI Second Strand Synthesis Module | --with_umi --umitools_extract_method "regex" --umitools_bc_pattern "^(?P<umi_1>.{6})(?P<discard_1>.{4}).*" |
| In sequence | Lexogen CORALL® Total RNA-Seq V1 > mind Appendix H regarding optional trimming |
--with_umi --umitools_extract_method "regex" --umitools_bc_pattern "^(?P<umi_1>.{12}).*"Optional: --clip_r2 9 --three_prime_clip_r2 12 |
| In sequence | Takara Bio SMART-Seq Total RNA Pico Input with UMIs and SMARTer® Stranded Total RNA-Seq Kit v3 | --with_umi --umitools_extract_method "regex" --umitools_bc_pattern2 "^(?P<umi_1>.{8})(?P<discard_1>.{6}).*" |
| In sequence | Watchmaker mRNA Library Prep Kit with Twist UMI Adapter System | --with_umi --umitools_extract_method "regex" --umitools_bc_pattern "^(?P<umi_1>.{5})(?P<discard_1>.{2}).*" --umitools_bc_pattern2 "^(?P<umi_2>.{5})(?P<discard_2>.{2}).*" |
No warranty for the accuracy or completeness of the parameters is implied
3′ digital gene expression assays
Some bulk RNA-seq library preparation protocols capture only a 3’ tag from each transcript, e.g. 3’Pool-seq, DRUG-seq, BRB-seq or Lexogen’s commercial QuantSeq 3’ mRNA-seq FWD protocol. The following parameters have been validated for QuantSeq 3' mRNA-seq FWD data, and provide useful starting points for other 3’ RNA-seq protocols:
Custom STAR parameters
Lexogen provides an example analysis workflow on their website, which includes the ENCODE standard options for the STAR aligner. In addition, Lexogen also decreases the tolerance for mismatches and clips poly(A) tails. To apply these settings, add the following parameters when running the pipeline:
--extra_star_align_args "--outFilterMismatchNoverLmax 0.1 --clip3pAdapterSeq AAAAAAAA"Note that many of the ENCODE standard STAR options (e.g. --outFilterMultimapNmax 20, --alignIntronMax 1000000) are already set as pipeline defaults. If you supply a parameter that duplicates a pipeline default, your value will take priority.
Custom Salmon arguments
Salmon’s default quantitation algorithm takes into account transcript length. Because 3’ tag protocols do not capture full transcripts, this feature needs to be deactivated by specifying:
--extra_salmon_quant_args "--noLengthCorrection"QuantSeq analysis with UMIs
If unique molecular identifiers were used to prepare the library, add the following arguments as well, to extract the UMIs and deduplicated alignments:
--with_umi--umitools_extract_method regex--umitools_bc_pattern "^(?P<umi_1>.{6})(?P<discard_1>.{4}).*"Reference genome options
Please refer to the nf-core website for general usage docs and guidelines regarding reference genomes.
Consistent reference resource usage
When supplying reference files as discussed below, it is important to be consistent in the reference resource used (Ensembl, GENCODE, UCSC etc), since differences in conventions between these resources can make their files incompatible. For example, UCSC prefixes chromosomes with chr, while Ensembl does not, so a GTF file from Ensembl should not be supplied alongside a genome FASTA from UCSC. GENCODE also attaches version identifiers to gene and transcript names (e.g. ENSG00000254647.1) while Ensembl does not.
Explicit reference file specification (recommended)
The minimum reference genome requirements for this pipeline are a FASTA file (genome and/ or transcriptome) and GTF file, all other files required to run the pipeline can be generated from these files. For example, the latest reference files for human can be derived from Ensembl like:
latest_release=$(curl -s 'http://rest.ensembl.org/info/software?content-type=application/json' | grep -o '"release":[0-9]*' | cut -d: -f2)wget -L ftp://ftp.ensembl.org/pub/release-${latest_release}/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa.gzwget -L ftp://ftp.ensembl.org/pub/release-${latest_release}/gtf/homo_sapiens/Homo_sapiens.GRCh38.${latest_release}.gtf.gzThese files can then be specified to the workflow with the --fasta and --gtf parameters.
Notes:
-
Compressed reference files are supported by the pipeline i.e. standard files with the
.gzextension and indices folders with thetar.gzextension. -
If
--gffis provided as input then this will be converted to a GTF file, or the latter will be used if both are provided. -
If
--gene_bedis not provided then it will be generated from the GTF file. -
If
--additional_fastais provided then the features in this file (e.g. ERCC spike-ins) will be automatically concatenated onto both the reference FASTA file as well as the GTF annotation before building the appropriate indices. Note: if you need the pipeline to build a pseudo-aligner index (Salmon/Kallisto),--additional_fastacannot be used together with--transcript_fastabecause the pipeline cannot append additional sequences to a user-provided transcriptome. Either omit--transcript_fastaand let the pipeline generate it, or provide a pre-built index that already contains the spike-ins. -
When using
--aligner star_rsem, the pipeline will build separate STAR and RSEM indices. STAR performs alignment with RSEM-compatible parameters, then RSEM quantifies from the resulting BAM files using--alignmentsmode. -
If the
--skip_alignmentoption is used along with--transcript_fasta, the pipeline can technically run without providing the genomic FASTA (--fasta). However, this approach is not recommended with--pseudo_aligner salmon, as any dynamically generated Salmon index will lack decoys. To ensure optimal indexing with decoys, it is highly recommended to include the genomic FASTA (--fasta) with Salmon, unless a pre-existing decoy-aware Salmon index is supplied. For more details on the benefits of decoy-aware indexing, refer to the Salmon documentation.
Reference genome
It is recommended to provide the most complete reference genome for your species, without additional loci (haplotypes) or patches. For model organisms such as mouse or human, this is the “primary assembly”, which includes the reference chromosomes and some additional scaffolds. For the human assembly GRCh38 (hg38), use the GRCh38.primary_assembly.genome.fa.gz file from GENCODE or the Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz file from Ensembl. These files cover the largest portion of the reference genome without including multiple copies of the same sequence, which would result in heavy mapping quality penalties.
For most other species (e.g., fly, cow, dog), no primary assembly is published. This reflects inadequately characterized genomic variation and a lower degree of curation, meaning that there are no established alternative loci (haplotypes), and that the toplevel file is equivalent to a primary assembly. Therefore, while the toplevel assembly may be utilized for these organisms, it is nonetheless advisable to verify the absence of N-padded haplotype or patch regions first.
Gene annotation
Gene annotations are updated more frequently than the reference genome sequence, so you must choose an appropriate annotation version (e.g. Ensembl release). We recommend using sources with well-defined, versioned releases such as ENSEMBL or GENCODE. Generally, it is best to use the most recent release for the latest gene annotations. However, if you are combining your data with older datasets, use the annotation version previously used for consistency.
Once you have chosen a release, select the annotation file that matches your reference genome. For the human primary assembly, use the comprehensive annotation (e.g., gencode.{release}.primary_assembly.annotation.gtf.gz from GENCODE or Homo_sapiens.GRCh38.{release}.gtf.gz from Ensembl). For other species, like fly, use the annotation matching the toplevel assembly (e.g., Drosophila_melanogaster.BDGP6.46.{release}.gtf.gz from Ensembl).
Ensure that the annotation files use gene IDs as the primary identifier, not the gene name/symbol. For example, the Ensembl ID ENSG00000254647 corresponds to the INS gene, which encodes the insulin protein. While gene names are more familiar, it is crucial to retain and use the primary identifiers as they are unique and easier to map between annotation versions or sources.
To take advantage of all the quality control modules implemented in the pipeline, the gene annotation should include a gene_biotype field which describes the function of each feature (protein coding, long non-coding etc.). This is usually the case for annotations from GENCODE or Ensembl but may not be if your annotation comes from another source. If your annotation does not include this field, please set the --skip_biotype_qc option to avoid running the steps that rely on it.
GTF vs GFF
GFF (General Feature Format) is a tab-separated text file format for representing genomic annotations, while GTF (General Transfer Format) is a specific implementation of this format corresponding to GFF version 2. The pipeline accepts both formats, but they must be provided via the correct parameter: use --gtf for GTF files (.gtf, .gtf.gz) and --gff for GFF3 files (.gff, .gff3, .gff.gz, .gff3.gz). GFF files provided via --gff will be automatically converted to GTF internally, so if a GTF is available for your annotation of choice it is better to provide that directly via --gtf.
More information and links to further resources are available from Ensembl.
Reference transcriptome
In addition to the reference genome sequence and annotation, you can provide a reference transcriptome FASTA file. These files can be obtained from GENCODE or Ensembl. However, these sequences only cover the reference chromosomes and can cause inconsistencies if you are using a primary or toplevel genome assembly and annotation.
We recommend not providing a transcriptome FASTA file and instead allowing the pipeline to create it from the provided genome and annotation. Similar to aligner indexes, you can save the created transcriptome FASTA and BED files to a central location for future pipeline runs. This helps avoid redundant computation and having multiple copies on your system. Ensure that all genome, annotation, transcriptome, and index versions match to maintain consistency.
If you are using --additional_fasta to add spike-in sequences (e.g. ERCC) and need the pipeline to build a pseudo-aligner index (Salmon/Kallisto), you must not provide --transcript_fasta. The pipeline needs to generate the transcriptome itself so that it includes the spike-in sequences. This combination will cause the pipeline to exit with an error unless you also provide a pre-built index (--salmon_index or --kallisto_index) that already contains the spike-in sequences.
Indices
By default, indices are generated dynamically by the workflow for tools such as STAR and Salmon. Since indexing is an expensive process in time and resources you should ensure that it is only done once, by retaining the indices generated from each batch of reference files by specifying --save_reference.
Once you have the indices from a workflow run you should save them somewhere central and reuse them in subsequent runs using custom config files or command line parameters such as --star_index '/path/to/STAR/index/'.
Remember to note the genome and annotation versions as well as the versions of the software used for indexing, as an index created with one version may not be compatible with other versions.
GENCODE
If you are using GENCODE reference genome files please specify the --gencode parameter because the format of these files is slightly different to ENSEMBL genome files:
- The
--featurecounts_group_typeparameter will automatically be set togene_typeas opposed togene_biotype, respectively. - If you are running Salmon, the
--gencodeflag will also be passed to the index building step to overcome parsing issues resulting from the transcript IDs in GENCODE fasta files being separated by vertical pipes (|) instead of spaces (see this issue).
As well as the standard annotations, GENCODE also provides “basic” annotations, which include only representative transcripts, but we do not recommend using these.
Prokaryotic genome annotations
The pipeline includes a dedicated profile for analysing bacterial and archaeal RNA-seq data:
This pipeline is primarily designed for eukaryotic RNA-seq. The prokaryotic profile provides basic support but does not include specialist features like operon detection or TSS mapping.
nextflow run nf-core/rnaseq \ --input samplesheet.csv \ --fasta genome.fasta \ --gff annotation.gff3 \ --outdir results \ -profile prokaryotic,dockerInput requirements
| Input | Parameter | Requirements |
|---|---|---|
| Samplesheet | --input |
Standard CSV format (see Samplesheet input) |
| Genome FASTA | --fasta |
Genomic sequence file (.fasta, .fa, .fna, optionally gzipped) |
| Annotation | --gff or --gtf |
Transcript-like features (CDS, tRNA, rRNA, tmRNA, ncRNA, etc.) with gene_id attributes. GFF3 format (.gff3, .gff) is typical for prokaryotes. |
Key points:
- Use GFF3 format: Prokaryotic annotations are typically distributed as GFF3 (not GTF). Provide GFF3 files via
--gffand GTF files via--gtf. - Transcripts are extracted from all transcript-like features: GFFREAD will extract transcript sequences from CDS, tRNA, rRNA, tmRNA, ncRNA and other feature types present in the annotation, so non-coding RNAs are quantified alongside mRNAs.
- CDS used for featureCounts QC:
featurecounts_feature_typeis set toCDS, so biotype/counting QC metrics are based on coding sequences. Your annotation should contain CDS features for these QC steps to be meaningful. - Matching contig names: Chromosome/contig names in your FASTA must exactly match those in your GFF/GTF (e.g., if your FASTA has
>NC_003197.2, your GFF must useNC_003197.2in column 1). - No transcript FASTA needed: The pipeline generates the transcript FASTA automatically using GFFREAD.
The -profile prokaryotic configures the pipeline with settings optimized for prokaryotic data:
- Bowtie2 alignment: Uses Bowtie2 instead of STAR as the default aligner. Prokaryotic genomes lack introns, so splice-aware aligners are unnecessary.
- GFFREAD transcript extraction: Uses GFFREAD instead of RSEM to generate transcript sequences. Prokaryotic annotations typically use CDS features rather than exon features, and RSEM requires exon features.
- CDS-based counting: Sets
featurecounts_feature_typetoCDSfor QC metrics. - Skips eukaryote-specific QC: Disables RSeQC and dupRadar, which assume eukaryotic gene structures.
You can override the aligner while keeping other prokaryotic settings:
# Use STAR instead of Bowtie2 with prokaryotic settingsnextflow run nf-core/rnaseq \ --input samplesheet.csv \ --fasta genome.fasta \ --gff annotation.gff3 \ --outdir results \ --aligner star_salmon \ -profile prokaryotic,dockerWhen using STAR with -profile prokaryotic, the pipeline automatically configures STAR to use CDS features instead of exons for both index building and alignment.
Annotation sources
For prokaryotic genomes, we recommend using annotations from Ensembl Bacteria when available, as these follow consistent formatting conventions. NCBI/RefSeq annotations can also work but may require additional attention:
- Contig name matching: Ensure chromosome/contig names in your FASTA match those in your GTF/GFF. This is a common source of errors when mixing files from different sources.
- Empty transcript IDs: Some bacterial GTFs from RefSeq contain gene entries with empty
transcript_idfields, which can cause Salmon errors. If you encounter issues, check your annotation file and consider filtering problematic entries.
Using GFFREAD independently
The --gffread_transcript_fasta parameter can also be used independently of -profile prokaryotic for any situation where RSEM fails to extract transcripts correctly (e.g., non-standard annotation formats):
nextflow run nf-core/rnaseq \ --input samplesheet.csv \ --fasta genome.fasta \ --gtf annotation.gtf \ --gffread_transcript_fasta \ --outdir results \ -profile dockerManual configuration (advanced)
If you prefer not to use the profile, you can manually configure the pipeline for prokaryotic data. The following parameters are set by -profile prokaryotic:
| Parameter | Value | Purpose |
|---|---|---|
--aligner |
bowtie2_salmon |
Splice-unaware alignment |
--gffread_transcript_fasta |
true |
Handle CDS-only annotations |
--featurecounts_feature_type |
CDS |
QC counting on CDS features |
--skip_rseqc |
true |
Skip eukaryote-specific QC |
--skip_dupradar |
true |
Skip eukaryote-specific QC |
--skip_qualimap |
true |
Skip eukaryote-specific QC |
--skip_bigwig |
true |
Inappropriate for transcriptomic alignments |
Additionally, you may want to set:
--skip_biotype_qc- if biotype information is not present or not of interest--skip_deseq2_qc- if you have fewer than 3 samples per condition
Please get in touch with us on the #rnaseq channel in the nf-core Slack workspace if you are having problems or need any advice.
Large chromosomes (plant genomes)
Genomes with very large chromosomes (>500 Mb), such as plant genomes, may encounter failures in the RSeQC inner_distance module due to a known limitation in the underlying bx-python library. The bx-python BitSet implementation has a maximum capacity of approximately 537 million bases, which can be exceeded by chromosomes in organisms like wheat, barley, and other plants.
If you encounter an error message similar to IndexError: [coordinate] is larger than the size of this BitSet (536870912), you can work around this by excluding the inner_distance module from RSeQC analysis:
--rseqc_modules 'bam_stat,infer_experiment,junction_annotation,junction_saturation,read_distribution,read_duplication'This removes inner_distance from the default list of RSeQC modules while retaining all other quality control metrics. Note that the inner_distance metric is only relevant for paired-end data and provides information about fragment size distribution.
For more information, see the upstream issues:
Reference catalogues via --genome
--genome <key> selects a bundle of reference paths from the params.genomes map and uses them in place of supplying --fasta/--gtf/etc. individually. The map is a generic catalogue mechanism: the pipeline ships the AWS iGenomes catalogue by default, but you can also author your own. iGenomes is the legacy use case; a user-maintained catalogue is the recommended way to get --genome ergonomics on modern reference data.
iGenomes (not recommended)
The pipeline ships the AWS iGenomes catalogue as the default params.genomes content. With nothing else configured, --genome GRCh37 (or any other iGenomes key) resolves to entries from this default catalogue, with paths pointing at AWS iGenomes (or the location specified by --igenomes_base if you’ve mirrored the catalogue locally). The bundled iGenomes STAR indices were built with an older STAR; the pipeline transparently adapts them so alignment runs with the modern STAR build the pipeline ships, no extra flags required.
If you override the resolved index by supplying your own --star_index, the pipeline assumes it is compatible with the modern STAR build.
The bundled iGenomes catalogue is provided for legacy compatibility but is not recommended for new analyses, because:
- Gene annotations in iGenomes are extremely out of date. This can be particularly problematic for RNA-seq analysis, which relies on accurate gene annotation.
- Some iGenomes references (e.g., GRCh38) point to annotation files that use gene symbols as the primary identifier. This can cause issues for downstream analysis, such as the nf-core differential abundance workflow where a conventional gene identifier distinct from symbol is expected.
For new analyses, supply --fasta/--gtf directly (see Explicit reference file specification above), or define your own catalogue using the same params.genomes shape (see below). A user-supplied catalogue can use whatever keys you want, including iGenomes-style names like GRCh37 to shadow the default entries with current reference data.
Custom catalogues
The params.genomes map is not iGenomes-specific. If you re-run the pipeline against a stable reference set, defining your own catalogue avoids the iGenomes downsides (stale annotations, version-locked STAR indices) while keeping the convenience of a single --genome <key> flag. Author a config file using the same map shape:
params { genomes { 'my_grch38_2025' { fasta = '/refs/grch38/genome.fa' gtf = '/refs/grch38/genes.gtf' transcript_fasta = '/refs/grch38/transcripts.fa' bed12 = '/refs/grch38/genes.bed' star = '/refs/grch38/star_2.7.11b/' salmon = '/refs/grch38/salmon/' } }}Run with -c my_genomes.config --genome my_grch38_2025. Any individual reference can still be overridden on the command line (e.g. --star_index /alternate/path).
The map keys are the bare names used internally (star, salmon, bed12, bowtie2, hisat2, kallisto, rsem, bbsplit, sortmerna, transcript_fasta, additional_fasta, gff, gtf, fasta); the pipeline maps these onto the corresponding --<name>_index parameters where appropriate.
Set star_legacy = true on an entry only if its star index was built with STAR 2.6.x (for example, an internal mirror of AWS iGenomes content). For indices built with modern STAR, leave the flag absent. The pipeline takes care of adapting flagged indices automatically; the flag has no effect if the user has overridden --star_index.
GTF filtering
By default, the input GTF file will be filtered to ensure that sequence names correspond to those in the genome fasta file (where supplied), and to remove rows with empty transcript identifiers. Filtering can be bypassed completely where you are confident it is not necessary, using the --skip_gtf_filter parameter. If you just want to skip the ‘transcript_id’ checking component of the GTF filtering script used in the pipeline this can be disabled specifically using the --skip_gtf_transcript_filter parameter.
Contamination screening options
The pipeline provides the option to scan reads for contamination from other species using either Sylph or Kraken2, with the possibility of applying corrections from Bracken. By default, contaminant screening runs on aligner-unmapped reads to preserve the existing pipeline behavior and focus on reads rejected by STAR or HISAT2. If you instead want to inspect the reads entering alignment, for example when evaluating the effect of BBSplit filtering, set --contaminant_screening_input trimmed. Since running Bracken is not computationally expensive, we recommend always using it to refine the abundance estimates generated by Kraken2.
Sylph is a faster and much more memory-efficient tool with about equal precision in species detection to Kraken2/Bracken. Sylph also has lower rates of false positives. However, Sylph does not assign specific reads to species; it only provides overall abundance estimates. Sylph abundance estimates also cannot assign a certain percentage of reads as unclassified.
Pre-constructed sylph databases can be found here and taxonomies here. The documentation also has instructions on creating custom databases/taxonomies. As a newer tool, the effect of database choice on Sylph’s performance has not been explored as thoroughly as for Kraken2 or Bracken. However, the following comments on choosing databases for Kraken2 are very likely still applicable to an extent for Sylph.
The accuracy of Kraken2 is highly dependent on the database used. Specifically, it is crucial to ensure that the host genome/transcriptome is included in the database. (Note that the pre-built sylph databases do not appear to contain the human genome/transcriptome). If you are particularly concerned about certain contaminants, it may be beneficial to use a smaller, more focused database containing primarily those contaminants instead of the full standard database. Various pre-built databases are available for download, and instructions for building a custom database can be found in the Kraken2 documentation. Additionally, genomes of contaminants detected in previous sequencing experiments are available on the OpenContami website.
While Kraken2 is capable of detecting low-abundance contaminants in a sample, false positives can occur. Therefore, if only a very small number of reads from a contaminating species are detected, these results should be interpreted with caution. Lastly, while Kraken2 can be used without Bracken, since running Bracken is not computationally expensive, we recommend always using it to refine the abundance estimates generated by Kraken2.
Running the pipeline
The typical command for running the pipeline is as follows:
nextflow run \ nf-core/rnaseq \ --input <SAMPLESHEET> \ --outdir <OUTDIR> \ --gtf <GTF> \ --fasta <GENOME FASTA> \ -profile dockerYou can also run without a genomic FASTA file, provided you skip the alignment step and provide a transcriptome FASTA directly:
nextflow run \ nf-core/rnaseq \ --input <SAMPLESHEET> \ --outdir <OUTDIR> \ --gtf <GTF> \ --transcript_fasta <TRANSCRIPTOME FASTA> \ --skip_alignment \ -profile dockerThis is not usually recommended with Salmon unless you also supply a previously generated decoy-aware Salmon transcriptome index.
The pipeline auto-loads the iGenomes catalogue config by default for consistency with other nf-core workflows. If you are not using iGenomes (e.g., supplying --fasta/--gtf directly or selecting a custom catalogue entry via --genome), set --igenomes_ignore to skip loading the bundled iGenomes map.
This will launch the pipeline with the docker configuration profile. See below for more information about profiles.
Note that the pipeline will create the following files in your working directory:
work # Directory containing the nextflow working files<OUTDIR> # Finished results in specified location (defined with --outdir).nextflow_log # Log file from Nextflow# Other nextflow hidden files, eg. history of pipeline runs and old logs.If you wish to repeatedly use the same parameters for multiple runs, rather than specifying each flag in the command, you can specify these in a params file.
Pipeline settings can be provided in a yaml or json file via -params-file <file>.
Do not use -c <file> to specify parameters as this will result in errors. Custom config files specified with -c must only be used for tuning process resource specifications, other infrastructural tweaks (such as output directories), or module arguments (args).
The above pipeline run specified with a params file in yaml format:
nextflow run nf-core/rnaseq -profile docker -params-file params.yamlwith:
input: <SAMPLESHEET>outdir: <OUTDIR>genome: 'GRCh37'<...>You can also generate such YAML/JSON files via nf-core/launch.
Running on Linux ARM architectures
Please note that the ARM profile is experimental. However, because testing is presently conducted manually, we cannot guarantee its reliability.
The pipeline can be executed in an ARM compatible mode by specifying the ARM profile, for example:
nextflow run \ nf-core/rnaseq \ --input <SAMPLESHEET> \ --outdir <OUTDIR> \ --gtf <GTF> \ --fasta <GENOME FASTA> \ -profile docker,arm64This will use ARM-compatible containers, and apply a small number of overrides to Conda definitions to support ARM operations.
Updating the pipeline
When you run the above command, Nextflow automatically pulls the pipeline code from GitHub and stores it as a cached version. When running the pipeline after this, it will always use the cached version if available - even if the pipeline has been updated since. To make sure that you’re running the latest version of the pipeline, make sure that you regularly update the cached version of the pipeline:
nextflow pull nf-core/rnaseqReproducibility
It is a good idea to specify the pipeline version when running the pipeline on your data. This ensures that a specific version of the pipeline code and software are used when you run your pipeline. If you keep using the same tag, you’ll be running the same version of the pipeline, even if there have been changes to the code since.
First, go to the nf-core/rnaseq releases page and find the latest pipeline version - numeric only (eg. 1.3.1). Then specify this when running the pipeline with -r (one hyphen) - eg. -r 1.3.1. Of course, you can switch to another version by changing the number after the -r flag.
This version number will be logged in reports when you run the pipeline, so that you’ll know what you used when you look back in the future. For example, at the bottom of the MultiQC reports.
To further assist in reproducibility, you can use share and reuse parameter files to repeat pipeline runs with the same settings without having to write out a command with every single parameter.
If you wish to share such profile (such as upload as supplementary material for academic publications), make sure to NOT include cluster specific paths to files, nor institutional specific profiles.
Core Nextflow arguments
These options are part of Nextflow and use a single hyphen (pipeline parameters use a double-hyphen)
-profile
Use this parameter to choose a configuration profile. Profiles can give configuration presets for different compute environments or analyses.
Several generic profiles are bundled with the pipeline which instruct the pipeline to use software packaged using different methods (Docker, Singularity, Podman, Shifter, Charliecloud, Apptainer, Conda) - see below.
We highly recommend the use of Docker or Singularity containers for full pipeline reproducibility, however when this is not possible, Conda is also supported.
The pipeline also dynamically loads configurations from https://github.com/nf-core/configs when it runs, making multiple config profiles for various institutional clusters available at run time. For more information and to check if your system is supported, please see the nf-core/configs documentation.
Note that multiple profiles can be loaded, for example: -profile test,docker - the order of arguments is important!
They are loaded in sequence, so later profiles can overwrite earlier profiles.
If -profile is not specified, the pipeline will run locally and expect all software to be installed and available on the PATH. This is not recommended, since it can lead to different results on different machines dependent on the computer environment.
test- A profile with a complete configuration for automated testing
- Includes links to test data so needs no other parameters
test_prokaryotic- A test profile for prokaryotic (bacterial/archaeal) RNA-seq data
- Uses Salmonella Typhimurium SL1344 test data with Bowtie2 alignment
- Includes all prokaryotic-specific settings; use
--aligner star_salmon --skip_bigwig falseto test with STAR
docker- A generic configuration profile to be used with Docker
singularity- A generic configuration profile to be used with Singularity
podman- A generic configuration profile to be used with Podman
shifter- A generic configuration profile to be used with Shifter
charliecloud- A generic configuration profile to be used with Charliecloud
apptainer- A generic configuration profile to be used with Apptainer
wave- A generic configuration profile to enable Wave containers. Use together with one of the above (requires Nextflow
24.03.0-edgeor later).
- A generic configuration profile to enable Wave containers. Use together with one of the above (requires Nextflow
conda- A generic configuration profile to be used with Conda. Please only use Conda as a last resort i.e. when it’s not possible to run the pipeline with Docker, Singularity, Podman, Shifter, Charliecloud, or Apptainer.
arm64- A configuration profile that applies overrides supplying ARM-compatible containers and Conda environments. See Running on Linux ARM architectures.
prokaryotic- A configuration profile optimized for bacterial and archaeal RNA-seq data. Uses Bowtie2 for alignment and configures the pipeline to handle CDS-based annotations. See Prokaryotic genome annotations.
-resume
Specify this when restarting a pipeline. Nextflow will use cached results from any pipeline steps where the inputs are the same, continuing from where it got to previously. For input to be considered the same, not only the names must be identical but the files’ contents as well. For more info about this parameter, see this blog post.
You can also supply a run name to resume a specific run: -resume [run-name]. Use the nextflow log command to show previous run names.
-c
Specify the path to a specific config file (this is a core Nextflow command). See the nf-core website documentation for more information.
Custom configuration
Resource requests
Whilst the default requirements set within the pipeline will hopefully work for most people and with most input data, you may find that you want to customise the compute resources that the pipeline requests. Each step in the pipeline has a default set of requirements for number of CPUs, memory and time. For most of the pipeline steps, if the job exits with any of the error codes specified here it will automatically be resubmitted with higher resources request (2 x original, then 3 x original). If it still fails after the third attempt then the pipeline execution is stopped.
To change the resource requests, please see the max resources and customise process resources section of the nf-core website.
Custom Containers
In some cases, you may wish to change the container or conda environment used by a pipeline steps for a particular tool. By default, nf-core pipelines use containers and software from the biocontainers or bioconda projects. However, in some cases the pipeline specified version maybe out of date.
To use a different container from the default container or conda environment specified in a pipeline, please see the updating tool versions section of the nf-core website.
Custom Tool Arguments
A pipeline might not always support every possible argument or option of a particular tool used in pipeline. Fortunately, nf-core pipelines provide some freedom to users to insert additional parameters that the pipeline does not include by default.
To learn how to provide additional arguments to a particular tool of the pipeline, please see the customising tool arguments section of the nf-core website.
nf-core/configs
In most cases, you will only need to create a custom config as a one-off but if you and others within your organisation are likely to be running nf-core pipelines regularly and need to use the same settings regularly it may be a good idea to request that your custom config file is uploaded to the nf-core/configs git repository. Before you do this please can you test that the config file works with your pipeline of choice using the -c parameter. You can then create a pull request to the nf-core/configs repository with the addition of your config file, associated documentation file (see examples in nf-core/configs/docs), and amending nfcore_custom.config to include your custom profile.
See the main Nextflow documentation for more information about creating your own configuration files.
If you have any questions or issues please send us a message on Slack on the #configs channel.
Running in the background
Nextflow handles job submissions and supervises the running jobs. The Nextflow process must run until the pipeline is finished.
The Nextflow -bg flag launches Nextflow in the background, detached from your terminal so that the workflow does not stop if you log out of your session. The logs are saved to a file.
Alternatively, you can use screen / tmux or similar tool to create a detached session which you can log back into at a later time.
Some HPC setups also allow you to run nextflow within a cluster job submitted your job scheduler (from where it submits more jobs).
Nextflow memory requirements
In some cases, the Nextflow Java virtual machines can start to request a large amount of memory.
We recommend adding the following line to your environment to limit this (typically in ~/.bashrc or ~./bash_profile):
NXF_OPTS='-Xms1g -Xmx4g'