nf-core/hlatyping
Precision HLA typing from next-generation sequencing data
1.1.5). The latest stable release is2.2.0.Introduction
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible.
Quick Start
If you want to test with a single line, if the pipeline works on your system, follow the next commands, with pre-configured test data-sets.
Docker
nextflow run nf-core/hlatyping -profile docker,test --outdir $PWD/resultsSingularity
nextflow run nf-core/hlatyping -profile singularity,test --outdir $PWD/resultsDocumentation
The nf-core/hlatyping pipeline comes with documentation about the pipeline, found in the docs/ directory:
- Installation
- Pipeline configuration
- Running the pipeline
- Output and how to interpret the results
- Troubleshooting
Pipeline DAG
The hlatyping pipeline can currently deal with two input formats: .fastq{.gz} or .bam, not both at the same time however. If the input file type is bam, than the pipeline extracts all reads from it and performs an mapping additional step with the yara mapper against the HLA reference sequence. Indices are provided in the ./data directory of this repository. Optitype uses razers3, which is very memory consuming. In order to avoid memory issues during pipeline execution, we reduce the mapping information on the relevant HLA regions on chromosome 6.
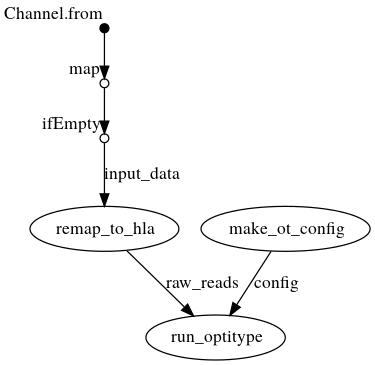
DAG with .fastq{.gz} as input
Creates a config file from the command line arguments, which is then passed to OptiType. In parallel, the fastqs are unzipped if they are passed as archives. OptiType is then used for the HLA typing.
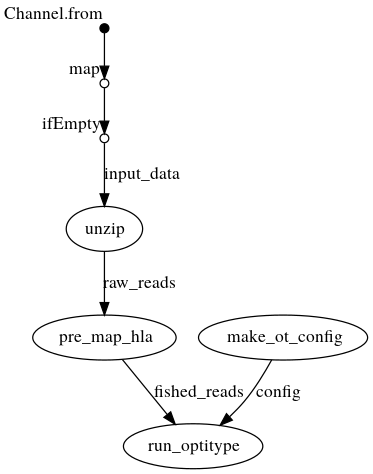
DAG with .bam as input
Creates a config file from the command line arguments, which is then passed to OptiType. In parallel, the reads are extracted from the bam file and mapped again against the HLA reference sequence on chromosome 6. OptiType is then used for the HLA typing.
Credits
This pipeline was originally written by: