nf-core/hlatyping
Precision HLA typing from next-generation sequencing data
1.2.0). The latest
stable release is
2.2.0
.
Introduction
The pipeline does next-generation sequencing-based Human Leukozyte Antigen (HLA) typing using OptiType. OptiType is a HLA genotyping algorithm based on integer linear programming. Reads of whole exome/genome/transcriptome sequencing data are mapped against a reference of known MHC class I alleles. To produce accurate 4-digit HLA genotyping predictions, all major and minor HLA-I loci are considered simultaneously to find an allele combination that maximizes the number of explained reads.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible.
Quick Start
-
Install
nextflow -
Install either
DockerorSingularityfor full pipeline reproducibility (please only useCondaas a last resort; see docs) -
Download the pipeline and test it on a minimal dataset with a single command:
nextflow run nf-core/hlatyping -profile test,<docker/singularity/conda/institute>Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment. -
Start running your own analysis!
nextflow run nf-core/hlatyping -profile <docker/singularity/conda/institute> --input '*_R{1,2}.fastq.gz'
See usage docs for all of the available options when running the pipeline.
Documentation
The nf-core/hlatyping pipeline comes with documentation about the pipeline which you can read at https://nf-co.re/hlatyping.
Pipeline DAG
The hlatyping pipeline can currently deal with two input formats: .fastq{.gz} or .bam, not both at the same time however. If the input file type is bam, than the pipeline extracts all reads from it and performs an mapping additional step with the yara mapper against the HLA reference sequence. Indices are provided in the ./data directory of this repository. Optitype uses razers3, which is very memory consuming. In order to avoid memory issues during pipeline execution, we reduce the mapping information on the relevant HLA regions on chromosome 6.
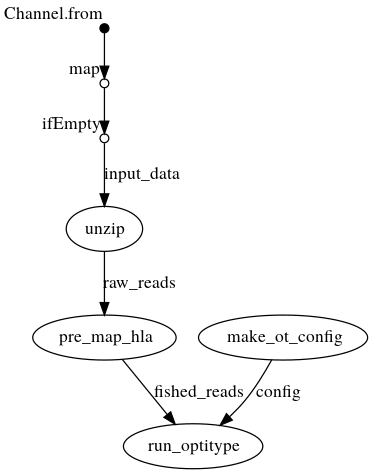
DAG with .fastq{.gz} as input
Creates a config file from the command line arguments, which is then passed to OptiType. In parallel, the fastqs are unzipped if they are passed as archives. OptiType is then used for the HLA typing.
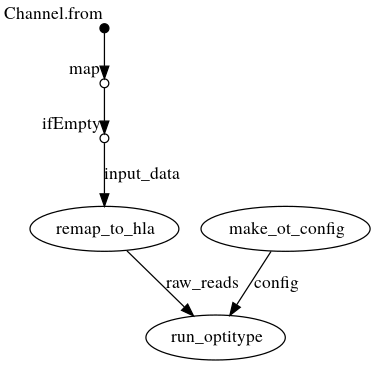
DAG with .bam as input
Creates a config file from the command line arguments, which is then passed to OptiType. In parallel, the reads are extracted from the bam file and mapped again against the HLA reference sequence on chromosome 6. OptiType is then used for the HLA typing.
Credits
nf-core/hlatyping was originally written by Christopher Mohr from Institute for Translational Bioinformatics and Quantitative Biology Center, Alexander Peltzer from Boeheringer Ingelheim, and Sven Fillinger from Quantitative Biology Center.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #hlatyping channel (you can join with this invite).
Citation
If you use nf-core/hlatyping for your analysis, please cite it using the following doi: 10.5281/zenodo.1401039
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x. ReadCube: Full Access Link