nf-core/mhcquant
Identify and quantify MHC eluted peptides from mass spectrometry raw data
2.0.0). The latest
stable release is
3.1.0
.
Introduction
nfcore/mhcquant is a bioinformatics analysis pipeline used for quantitative processing of data dependent (DDA) peptidomics data.
It was specifically designed to analyse immunopeptidomics data, which deals with the analysis of affinity purified, unspecifically cleaved peptides that have recently been discussed intensively in the context of cancer vaccines.
The workflow is based on the OpenMS C++ framework for computational mass spectrometry. RAW files (mzML) serve as inputs and a database search (Comet) is performed based on a given input protein database. FDR rescoring is applied using Percolator based on a competitive target-decoy approach (reversed decoys). For label free quantification all input files undergo identification based retention time alignment (MapAlignerIdentification), and targeted feature extraction matching ids between runs (FeatureFinderIdentification). In addition, a variant calling file (vcf) can be specified to translate variants into proteins that will be included in the database search and binding predictions on specified alleles (alleles.tsv) using MHCFlurry (Class 1) or MHCNugget (Class 2) can be directly run on the output peptide lists. Moreover, if a vcf file was specified, neoepitopes will automatically be determined and binding predictions can also directly be predicted for them.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible.
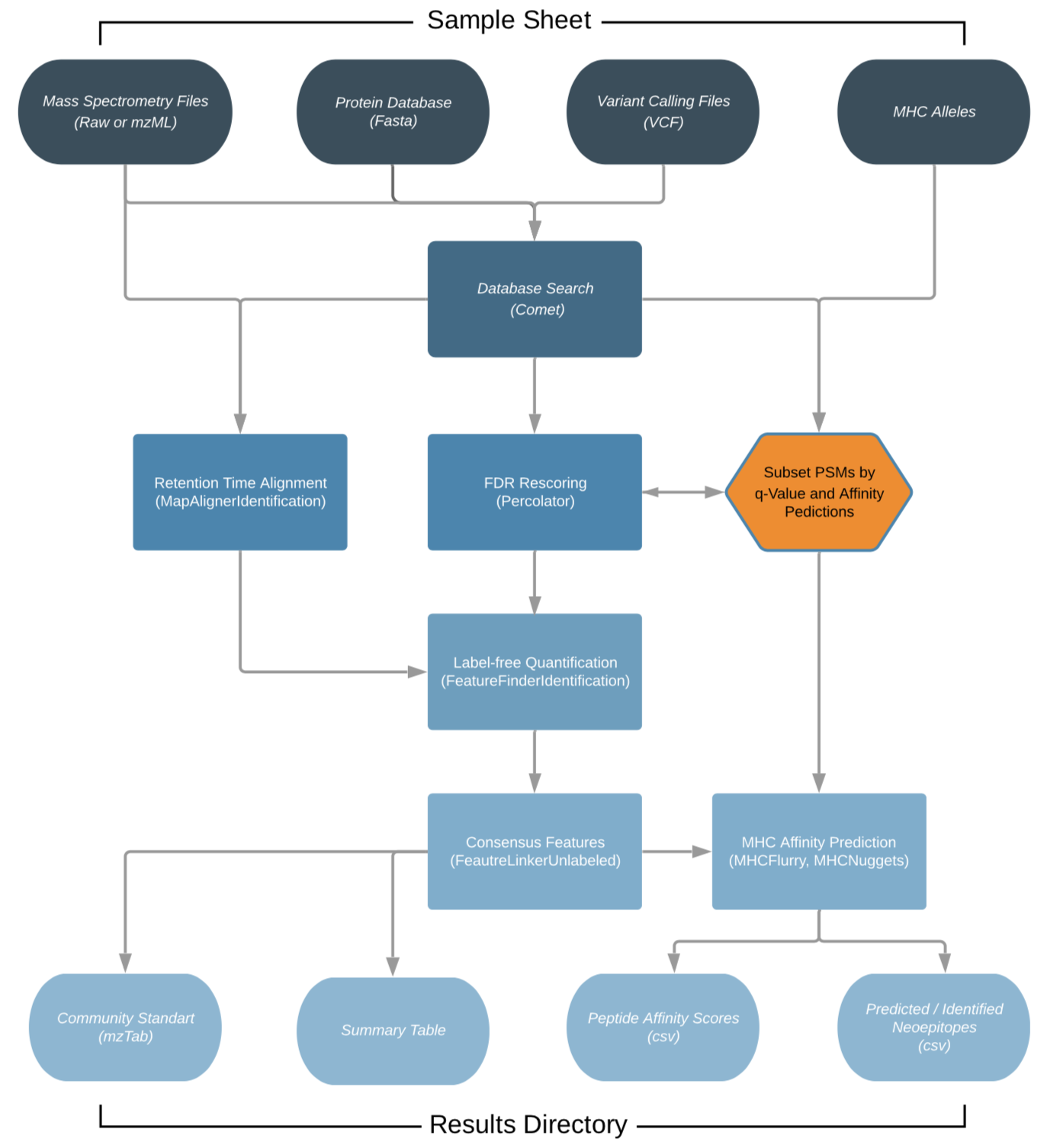 (This chart was created with the help of Lucidchart)
(This chart was created with the help of Lucidchart)
Quick Start
-
Install
nextflow(>=21.04.0) -
Install any of
Docker,Singularity,Podman,ShifterorCharliecloudfor full pipeline reproducibility (please only useCondaas a last resort; see docs) -
Download the pipeline and test it on a minimal dataset with a single command:
nextflow run nf-core/mhcquant -profile test,<docker/singularity/podman/shifter/charliecloud/conda/institute>Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment. -
Start running your own analysis!
nextflow run nf-core/mhcquant -profile test,<docker/singularity/podman/shifter/charliecloud/conda/institute> --input 'samples.tsv' --fasta 'SWISSPROT_2020.fasta' --allele_sheet 'alleles.tsv' --predict_class_1 --refine_fdr_on_predicted_subset
See usage docs for all of the available options when running the pipeline.
Pipeline Summary
By default, the pipeline currently performs the following:
- Protein database addition by mutated genome variants (
Fred2 Immunoinformatics Toolbox) - Database search (‘Comet’)
- False discovery rate estimation (‘Percolator’)
- Retention time alignment (‘OpenMS-MapAlignerIdentification’)
- Targeted peptide quantification (‘OpenMS-FeatureFinderIdentification’)
- MHC peptide affinity prediction (‘MHCFlurry’,‘MHCNuggets’)
Documentation
The nf-core/mhcquant pipeline comes with documentation about the pipeline: usage and output.
Credits
We thank the following people for their extensive assistance in the development of this pipeline:
- Leon Bichmann (@Leon-Bichmann)
- Lukas Heumos (@Zethson)
- Alexander Peltzer (@apeltzer)
The pipeline was converted to Nextflow DSL2 by Marissa Dubbelaar (@marissaDubbelaar)
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #mhcquant channel (you can join with this invite).
Citations
If you use nf-core/mhcquant for your analysis, please cite:
MHCquant: Automated and Reproducible Data Analysis for Immunopeptidomics
Leon Bichmann, Annika Nelde, Michael Ghosh, Lukas Heumos, Christopher Mohr, Alexander Peltzer, Leon Kuchenbecker, Timo Sachsenberg, Juliane S. Walz, Stefan Stevanović, Hans-Georg Rammensee & Oliver Kohlbacher
Journal of Proteome Research 2019 18 (11), 3876-3884 DOI: 10.1021/acs.jproteome.9b00313
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
In addition, references of tools and data used in this pipeline are as follows:
Fred2 Immunoinformatics Toolbox
Schubert B. et al, Bioinformatics 2016 Jul 1;32(13):2044-6. doi: 10.1093/bioinformatics/btw113. Epub 2016 Feb 26
Comet Search Engine
Eng J.K. et al, J Am Soc Mass Spectrom. 2015 Nov;26(11):1865-74. doi: 10.1007/s13361-015-1179-x. Epub 2015 Jun 27.
Percolator
Käll L. et al, Nat Methods 2007 Nov;4(11):923-5. doi: 10.1038/nmeth1113. Epub 2007 Oct 21.
Identification based RT Alignment
Weisser H. et al, J Proteome Res. 2013 Apr 5;12(4):1628-44. doi: 10.1021/pr300992u. Epub 2013 Feb 22.
Targeted peptide quantification
Weisser H. et al, J Proteome Res. 2017 Aug 4;16(8):2964-2974. doi: 10.1021/acs.jproteome.7b00248. Epub 2017 Jul 19.
MHC affinity prediction
O’Donnell T.J., Cell Syst. 2018 Jul 25;7(1):129-132.e4. doi: 10.1016/j.cels.2018.05.014. Epub 2018 Jun 27.
Shao X.M., Cancer Immunol Res. 2020 Mar;8(3):396-408. doi: 10.1158/2326-6066.CIR-19-0464. Epub 2019 Dec 23.