nf-core/omicsgenetraitassociation
A nextflow pipeline which integrates multiple omic data streams and performs coordinated analysis
Introduction
nf-core/omicsgenetraitassociation is a bioinformatics pipeline that can be used to perform meta-analysis of trait associations accounting for correlations across omics studies due to hidden non-independencies between study elements which may arise from overlapping or related samples. It takes a samplesheet with input omic association data, performs gene-level aggregation, correlated meta-analysis, and produces a report on downstream module enrichment and gene ontology enrichment analyses.
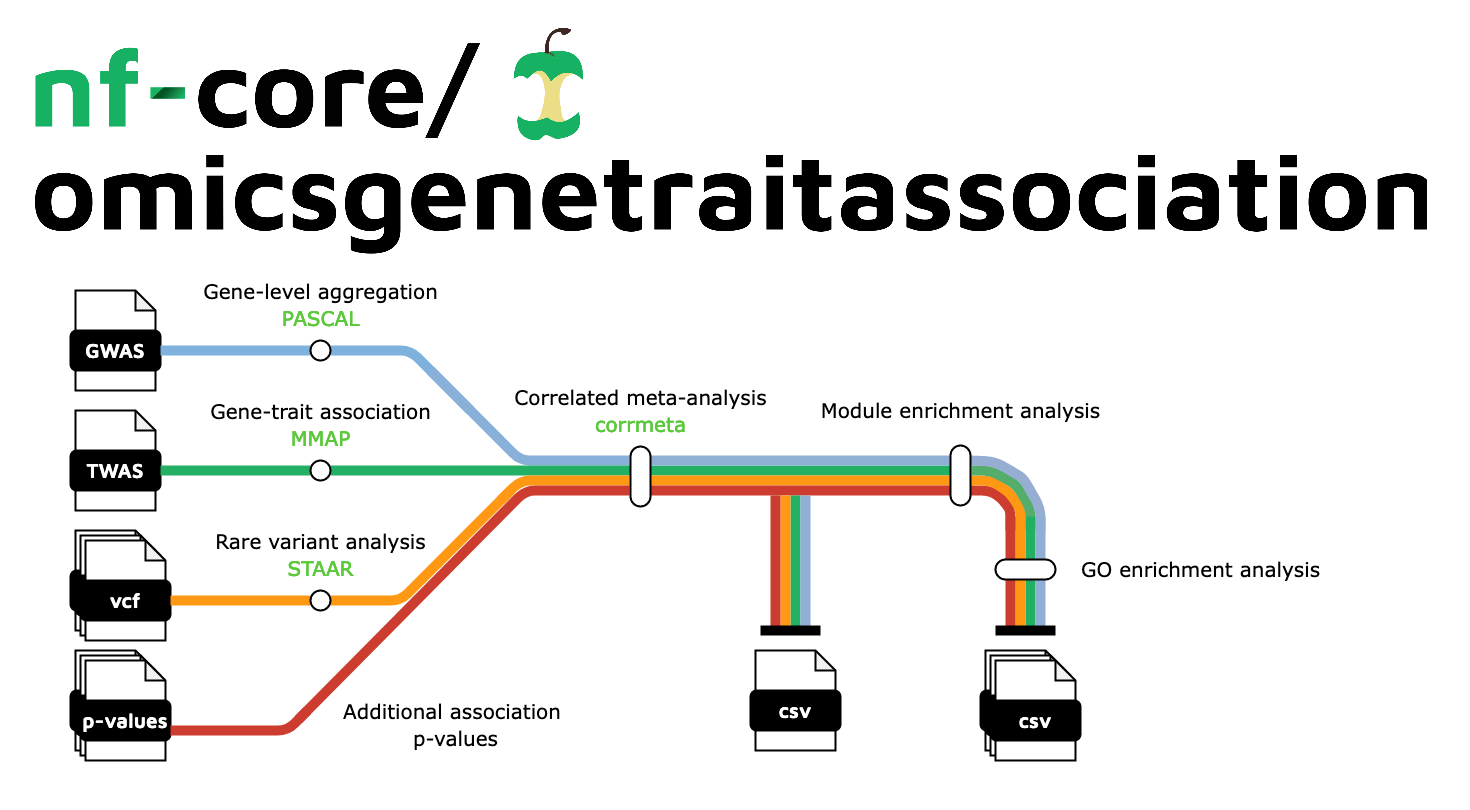
- Gene-level aggregation of GWAS summary statistics
PASCAL - Gene-trait association
MMAP - Correlated meta-analysis
corrmeta - Module enrichment analysis
MEA - Gene ontology (GO) enrichment analysis
GO
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,trait,pascal,twas,additional_sourcesllfs_fhshdl,fhshdl,data/llfs/fhshdl/gwas.csv,data/llfs/fhshdl/twas.csv,data/llfs/additional_sources.txtfhs_lnTG,lnTG,data/fhs/lnTG/gwas.csv,data/fhs/lnTG/twas.csv,Each row represents a single correlated meta-analysis run. pascal is the GWAS summary statistics to be aggreagted to the gene-level. twas is the gene-trait association phenotype file (please refer to usage.md for details). additional_sources lists paths to additional omic association p-values.
Now, you can run the pipeline using:
nextflow run nf-core/omicsgenetraitassociation \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/omicsgenetraitassociation was originally written by Woo Jung (@wsjung).
Many thanks to others who have written parts of the pipeline or helped out along the way too, including (but not limited to):
- Chase Mateusiak
- Sandeep Acharya
- Edward Kang
- Lisa Liao
- Michael Brent
- Michael Province
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #omicsgenetraitassociation channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.