nf-core/riboseq
Pipeline for the analysis of ribosome profiling, or Ribo-seq (also named ribosome footprinting) data.
Introduction
nf-core/riboseq is a bioinformatics pipeline for analysis of Ribo-seq data. It borrows heavily from nf-core/rnaseq in the preprocessing stages:
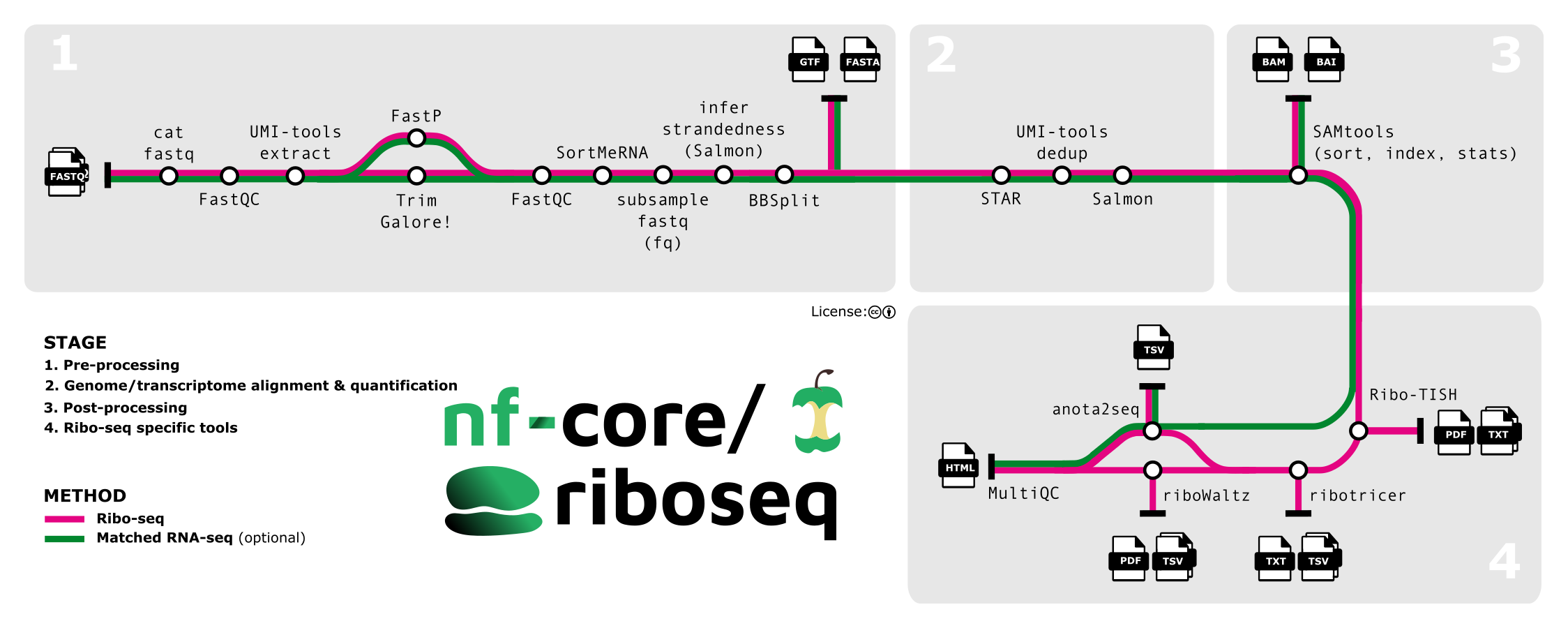
- Merge re-sequenced FastQ files (
cat) - Sub-sample FastQ files and auto-infer strandedness (
fq,Salmon) - Read QC (
FastQC) - UMI extraction (
UMI-tools) - Adapter and quality trimming (
Trim Galore!) - Removal of genome contaminants (
BBSplit) - Removal of ribosomal RNA (
SortMeRNA) - Genome alignment of reads, outputting both genome and transcriptome alignments with
STAR - Sort and index alignments (
SAMtools) - UMI-based deduplication (
UMI-tools)
Differences occur in the downstream analysis steps. Currently these specialist steps are:
- Check reads distribution around annotated protein coding regions on user provided transcripts, show frame bias and estimate P-site offset for different group of reads (
Ribo-TISH) - (default, optional) Predict translated open reading frames and/ or translation initiation sites de novo from alignment data (
Ribo-TISH) - (opt-in,
--run_ribotricer) Derive candidate ORFs from reference data and detect translated ORFs from that list (Ribotricer). Disabled by default: benchmarking found its ORF-score column is rank-unstable across biological replicates, so it is excluded from the default caller set and from cross-caller rank aggregation when enabled. - (default, optional) Identify translated ORFs using P-site periodicity and read density (
RiboCode) - (default, optional) Derive P-sites and QC from transcriptome alignments (
riboWaltz,plastid) - (optional) Use a translational efficiency approach to study the dynamics of transcription and translation, with anota2seq. requires matched RNA-seq and Ribo-seq data
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,fastq_1,fastq_2,strandedness,typeCONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,forward,riboseqEach row represents a fastq file (single-end) or a pair of fastq files (paired end). Each row should have a ‘type’ value of riboseq, tiseq or rnaseq. Future iterations of the workflow will conduct paired analysis of matched riboseq and rnaseq samples to accomplish analysis types such as ’translational efficiency, but in the current version you should set this to riboseq or tiseq for reglar Ribo-seq or TI-seq data respectively.
Now, you can run the pipeline using:
nextflow run nf-core/riboseq \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --outdir <OUTDIR>Including a translational efficiency analysis
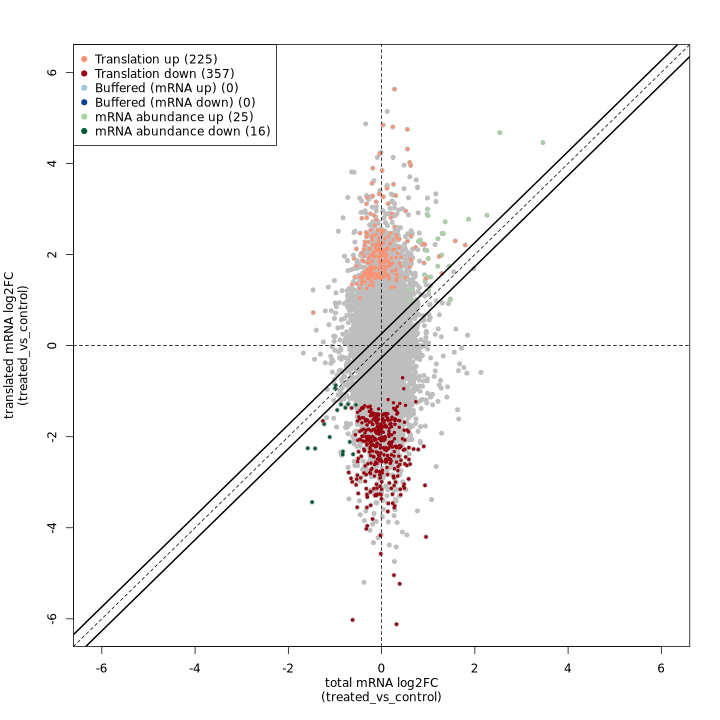
In the translational efficiency analysis provided by anota2seq, we use matched pairs of Ribo-seq and RNA-seq data to study the relationship between transcription and translation as they differ between two treatment groups. For example the test data for this workflow has a contrasts file like:
id,variable,reference,target,batch,pairtreated_vs_control,treatment,control,treated,,pairThis describes how to compare groups of samples between treament groups, and between RNA-seq and Ribo-seq. In order the columns are:
id: a unique identifier to use for the contrast- ’variable`: which vaiable (column) of the sample sheet should be used to separate the treatment groups?
reference: which value of the variable column should be used to select samples to be used as the reference/ base group?target: which value of the variable column should be used to select samples to be used as the target/treated group?batch: (optional) specify a variable in the sample sheet that defines sample batchespair: (optional) specify a variable in the sample sheet that defines sample pairing between RNA-seq and Ribo-seq samples. If not specified, it is assumed that the two types of sample are ordered the same.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/riboseq was originally written by Jonathan Manning (Bioinformatics Engineer at Seqera) with support from Altos Labs and in discussion with Felix Krueger and Christel Krueger. We thank the following people for their input:
- Anne Bresciani (ZS)
- Felipe Almeida (ZS)
- Mikhail Osipovitch (ZS)
- Edward Wallace (University of Edinburgh)
- Jack Tierney (University College Cork)
- Maxime U Garcia (Seqera)
- Ira A Iosub (The Francis Crick Institute)
- Sebastian Uhrig (Freelance bioinformatician)
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #riboseq channel (you can join with this invite).
Citations
If you use nf-core/riboseq for your analysis, please cite it using the following doi: 10.5281/zenodo.10966364
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.