nf-core/ampliseq
Amplicon sequencing analysis workflow using DADA2 and QIIME2
Introduction
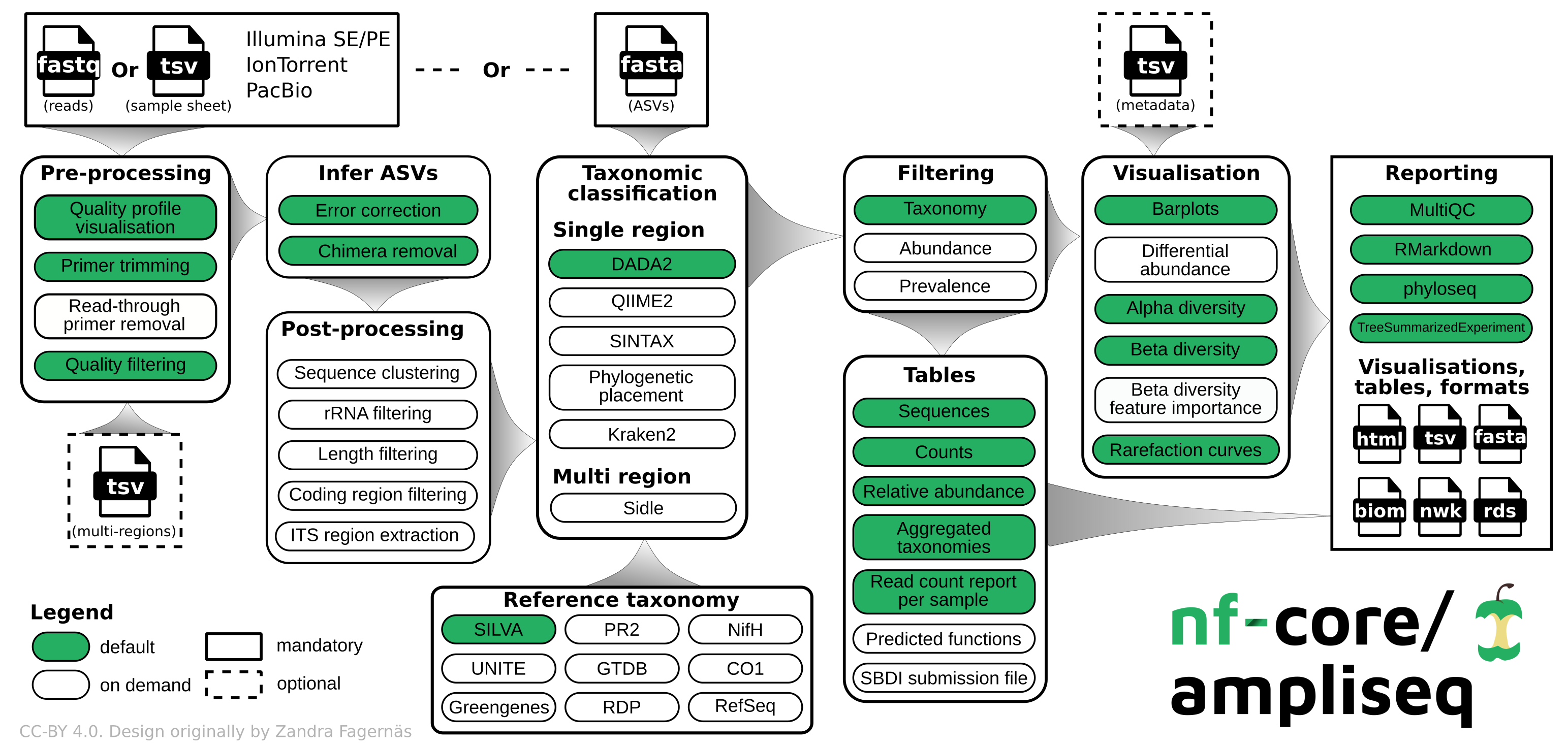
nfcore/ampliseq is a bioinformatics analysis pipeline used for amplicon sequencing, supporting denoising of any amplicon and supports a variety of taxonomic databases for taxonomic assignment including 16S, ITS, CO1 and 18S. Phylogenetic placement is also possible. Multiple region analysis such as 5R is implemented. Supported is paired-end Illumina or single-end Illumina, PacBio, IonTorrent, and Oxford Nanopore data. Default is the analysis of 16S rRNA gene amplicons sequenced paired-end with Illumina.
A video about relevance, usage and output of the pipeline (version 2.1.0; 26th Oct. 2021) can also be found in YouTube and billibilli, the slides are deposited at figshare.
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Pipeline summary
By default, for paired-end Illumina data, the pipeline currently performs the following:
- Sequencing quality control (FastQC)
- Trimming of reads (Cutadapt)
- Infer Amplicon Sequence Variants (ASVs) (DADA2)
- Optional post-clustering with VSEARCH
- Decontamination with decontam, if any controls or quantification information is given
- Predict whether ASVs are ribosomal RNA sequences (Barrnap)
- Phylogenetic placement (EPA-NG)
- Taxonomical classification using DADA2; alternatives are SINTAX, Kraken2, and QIIME2
- Excludes unwanted taxa, produces absolute and relative feature/taxa count tables and plots, plots alpha rarefaction curves, computes alpha and beta diversity indices and plots thereof (QIIME2)
- Creates phyloseq R objects (Phyloseq and TreeSE)
- Pipeline QC summaries (MultiQC)
- Pipeline summary report (R Markdown)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, you need to know whether the sequencing files at hand are expected to contain primer sequences (usually yes) and if yes, what primer sequences. In the example below, the paired end sequencing data was produced with 515f (GTGYCAGCMGCCGCGGTAA) and 806r (GGACTACNVGGGTWTCTAAT) primers of the V4 region of the 16S rRNA gene. Please note, that those primer sequences should not contain any sequencing adapter sequences, only the sequence that matches the biological amplicon.
Next, the sequencing data needs to be detailed in a sample sheet or organized in a folder (see input documentation).
Furthermore, specify what type of sequencing data you have, e.g. Illumina, PacBio, IonTorrent, and Oxford Nanopore data (see data type documentation). In the example below paired-end Illumina data is assumed, which is default.
Now, you can run the pipeline using:
nextflow run nf-core/ampliseq \ -profile <docker/singularity/.../institute> \ --input "samplesheet.tsv" \ --primer_fwd "GTGYCAGCMGCCGCGGTAA" \ --primer_rev "GGACTACNVGGGTWTCTAAT" \ --outdir <OUTDIR>Adding metadata will considerably increase the output, see metadata documentation.
By default the taxonomic assignment will be performed with DADA2 on SBDI-GTDB database, but there are various tools and databases readily available, see taxonomic classification documentation.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/ampliseq was originally written by Daniel Straub (@d4straub) and Alexander Peltzer (@apeltzer) for use at the Quantitative Biology Center (QBiC) and Microbial Ecology, Center for Applied Geosciences, part of Eberhard Karls Universität Tübingen (Germany). Daniel Lundin @erikrikarddaniel (Linnaeus University, Sweden) joined before pipeline release 2.0.0 and helped to improve the pipeline considerably.
We thank the following people for their extensive assistance in the development of this pipeline (in alphabetical order):
Adam Bennett, Diego Brambilla, Emelie Nilsson, Jeanette Tångrot, Lokeshwaran Manoharan, Marissa Dubbelaar, Sabrina Krakau, Sam Minot, Till Englert
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #ampliseq channel (you can join with this invite).
Citations
If you use nf-core/ampliseq for your analysis, please cite the ampliseq article as follows:
Interpretations of Environmental Microbial Community Studies Are Biased by the Selected 16S rRNA (Gene) Amplicon Sequencing Pipeline
Daniel Straub, Nia Blackwell, Adrian Langarica-Fuentes, Alexander Peltzer, Sven Nahnsen, Sara Kleindienst
Frontiers in Microbiology 2020, 11:2652 doi: 10.3389/fmicb.2020.550420.
You can cite the nf-core/ampliseq zenodo record for a specific version using the following doi: 10.5281/zenodo.1493841
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.