nf-core/bactmap
A mapping-based pipeline for creating a phylogeny from bacterial whole genome sequences
Introduction
nf-core/bactmap is a bioinformatics best-practice analysis pipeline for mapping short (Illumina) and long reads (Oxford Nanopore) from bacterial WGS to a reference sequence, creating filtered VCF files and making pseudogenomes based on high quality positions in the VCF files.
Pipeline summary
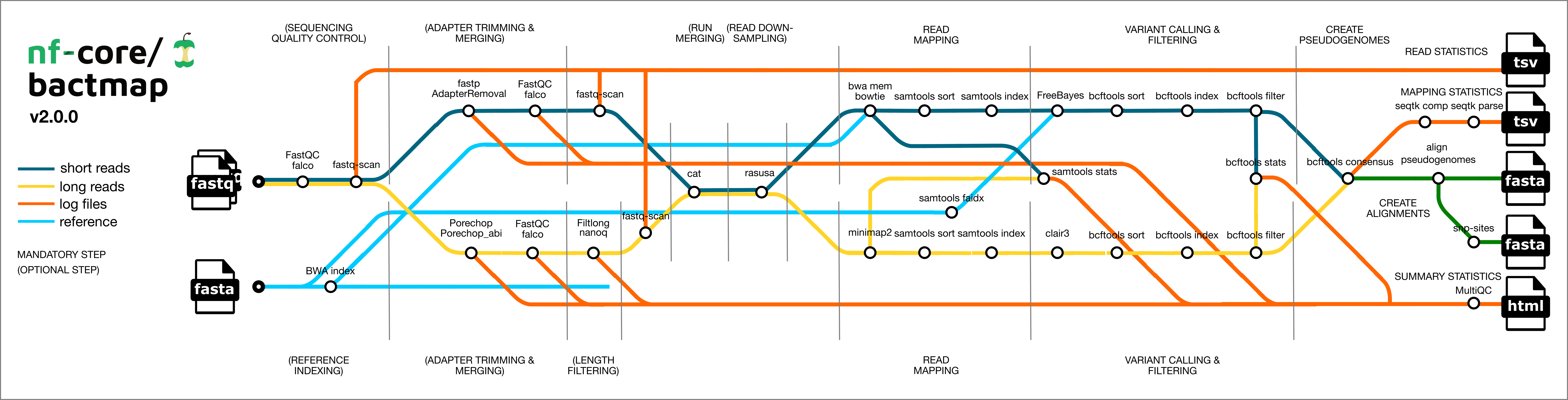
- Index reference fasta file (short-read:
BWA indexorBowtie2 build; long-read:minimap2 index) - Read QC (
FastQCorfalcoas an alternative option) - Calculate fastq summary statistics (
fastq-scan) - Perform read pre-processing (optional)
- Adapter clipping and merging (short-read:
fastporAdapterRemoval2; long-read:porechoporPorechop_ABI) - Quality filtering (long-read:
Filtlong),Nanoq - Run merging (
cat)
- Adapter clipping and merging (short-read:
- Downsample fastq files (optional) (
Rasusa) - Summarise read statistics pre- and post-processing and subsampling (
read_stats) - Variant calling
- Map reads to reference (short-read:
BWA-MEM2orBowtie2; long-read:minimap2) - Sort and index alignments (
SAMtools view/sort) - Summarise alignment statistics (
SAMtools stats) - Call variants (short-read:
FreeBayes; long-read:Clair3) - Filter variants (
BCFtools filter) - Summarise variant statistics (
BCFtools stats) - Convert filtered bcf to pseudogenome fasta (
BCFtools consensusandBEDtools) - Summarise mapping statistics (
seqtk)
- Create alignment from pseudogenomes by concatenating fasta files having first checked that the sample sequences are high quality (
alignpseudogenomes) - Extract variant sites from alignment (
SNP-sites) - Present QC for raw and processed reads, alignment statistics and variant statistics (
MultiQC)
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
sample,run_accession,instrument_platform,fastq_1,fastq_22612,run1,ILLUMINA,2612_run1_R1.fq.gz,2613,run1,ILLUMINA,2612_run3_R1.fq.gz,2612_run3_R2.fq.gz2614,run3,OXFORD_NANOPORE,2614_file1.fastq.gz,2614,run3,OXFORD_NANOPORE,2614_file2.fastq.gz,Each row represents a fastq file (single-end) or a pair of fastq files (paired end), either Illumina (short reads) or Oxford Nanopore (long reads).
Additionally, if you are analysing Oxford Nanopore data, you will need to provide the path to a model to use with Clair3 (specified with --clair3_model). Models for older chemistries and basecallers (e.g. r9.4.1) can be downloaded from here. For newer chemistries and basecallers, ONT provides models through Rerio. To download the models for Clair3 from the ONT github, you can use the following commands (each model will be downloaded to the folder clair3_models/<clair3_model_name>):
# Clone the rerio repositorygit clone https://github.com/nanoporetech/rerio
# Download all modelspython3 download_model.py --clair3Now, you can run the pipeline using:
nextflow run nf-core/bactmap \ -profile <docker/singularity/.../institute> \ --input samplesheet.csv \ --fasta <REFERENCE_FASTA> \ --clair3_model <PATH_TO_CLAIR3_MODEL> \ --outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/bactmap was originally written by Anthony Underwood, Andries van Tonder and Thanh Le Viet.
We thank the following people for their extensive assistance in the development of this pipeline:
- Alexandre Gilardet
- Hanh Hoang
- Ismael Henarejos-Castilo
- Mareike Janiak
- Harshil Patel
- Olha Petryk
- Richard Agyekum
- Steven Sutcliffe
- Szymon Szyszkowski
Anthony Underwood’s time working on the project was funded by the National Institute for Health Research(NIHR) Global Health Research Unit for the Surveillance of Antimicrobial Resistance (Grant Reference Number 16/136/111)

Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #bactmap channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.