nf-core/mcmicro
An end-to-end processing pipeline that transforms multi-channel whole-slide images into single-cell data.
Introduction
We are currently in the process of porting the original MCMICRO to nf-core. This pipeline is therefore in active development.
nf-core/mcmicro is a nextflow pipeline for processing highly-multiplexed imaging data, as produced by technologies such as Cycif, MIBI, CODEX, SeqIF among others.
If you want to run the original MCMICRO pipeline outside of nf-core, please see https://mcmicro.org/.
The nf-core/mcmicro pipeline is an end-to-end processing pipeline that transforms multi-channel whole-slide images into single-cell data. It takes samplesheet and markersheet files as input and perfoms registration, segmentation and quantification. Multiple segmentation modules are available and can be run in parallel. The pipeline can also optionally perform background and shading correction, background subtraction, and supports tissue microarrays. It returns a pre-segmentation image file, a segmentation mask image, and a cell x feature array spreadsheet.
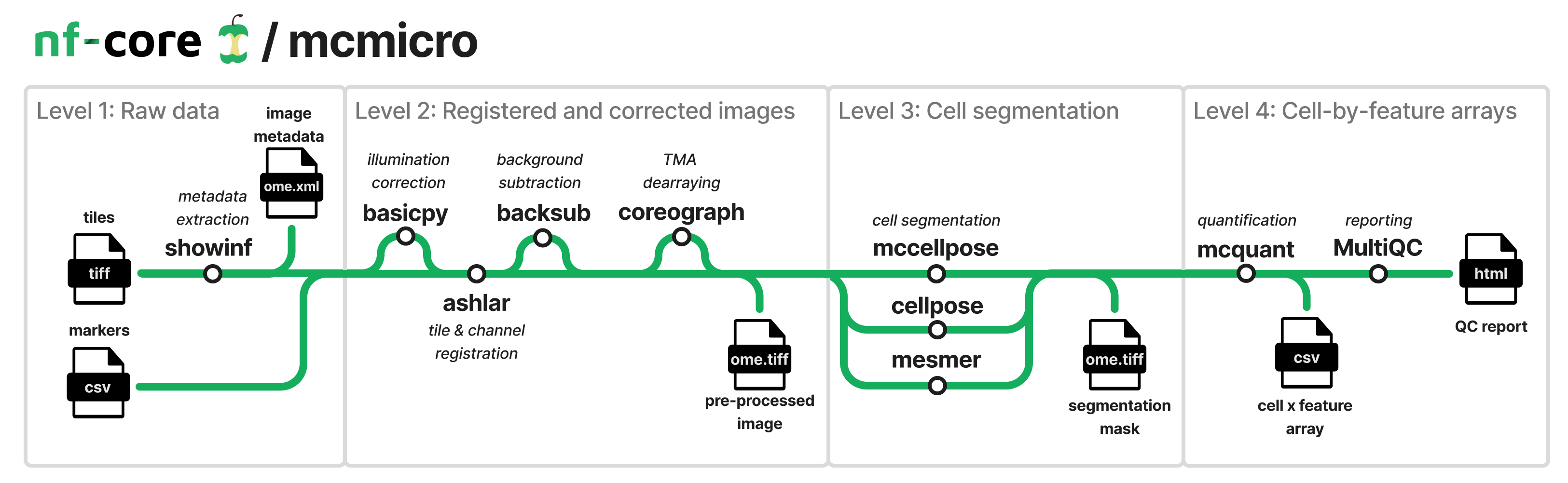
The MC in MCMICRO stands for ‘Multiple Choice’ and that refers to the fact that there will be multiple module options available for many steps in the pipeline. The currently supported options are provided in parentheses for required steps.
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet and markersheet with your input data that looks as follows:
samplesheet.csv:
sample,cycle_number,image_tiles
TEST1,1,https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/imaging/ome-tiff/cycif-tonsil-cycle1.ome.tif
Each row of the samplesheet represents a single cycle ome.tiff microscopy image file. The first column sample is the sample identifier. It can be repeated in multiple rows for samples with multiple cycles. The second column cycle_number is a number corresponding to the cycle number of the image. The third column image_tiles is a path or URL to the input image file.
markersheet.csv:
channel_number,cycle_number,marker_name
1,1,DNA 1
2,1,Na/K ATPase
3,1,CD3
4,1,CD45ROEach row of the markersheet represents a single channel in the associated sample image. The first column channel_number is an identifier for the respective channel. The second column cycle_number corresponds to the cycle number of the image and it must match the cycle_number in the supplied samplesheet. The third column marker_name is the name of the marker for the given channel and cycle.
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
Now, you can run the pipeline using:
nextflow run nf-core/mcmicro \
-profile <docker/singularity/.../institute> \
--input_cycle samplesheet.csv \
--marker_sheet markers.csv \
--outdir <OUTDIR>For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/mcmicro was written by Rob Young and Jeremy Muhlich from the Laboratory of Systems Pharmacology at Harvard Medical School. It is based on version 1.0 of mcmicro (labsyspharm/mcmicro) which was also developed there.
We thank the following people for their assistance in the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #mcmicro channel (you can join with this invite).
Citations
If you use nf-core/mcmicro for your analysis, please cite it using the following article: Schapiro et al. 2022 Nat. Methods
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.