nf-core/mhcquant
Identify and quantify MHC eluted peptides from mass spectrometry raw data
Introduction
nfcore/mhcquant is a best-practice bioinformatics pipeline to process data-dependent acquisition (DDA) immunopeptidomics data. This involves mass spectrometry-based identification and quantification of immunopeptides presented on major histocompatibility complex (MHC) molecules which mediate T cell immunosurveillance. Immunopeptidomics has central implications for clinical research, in the context of T cell-centric immunotherapies.
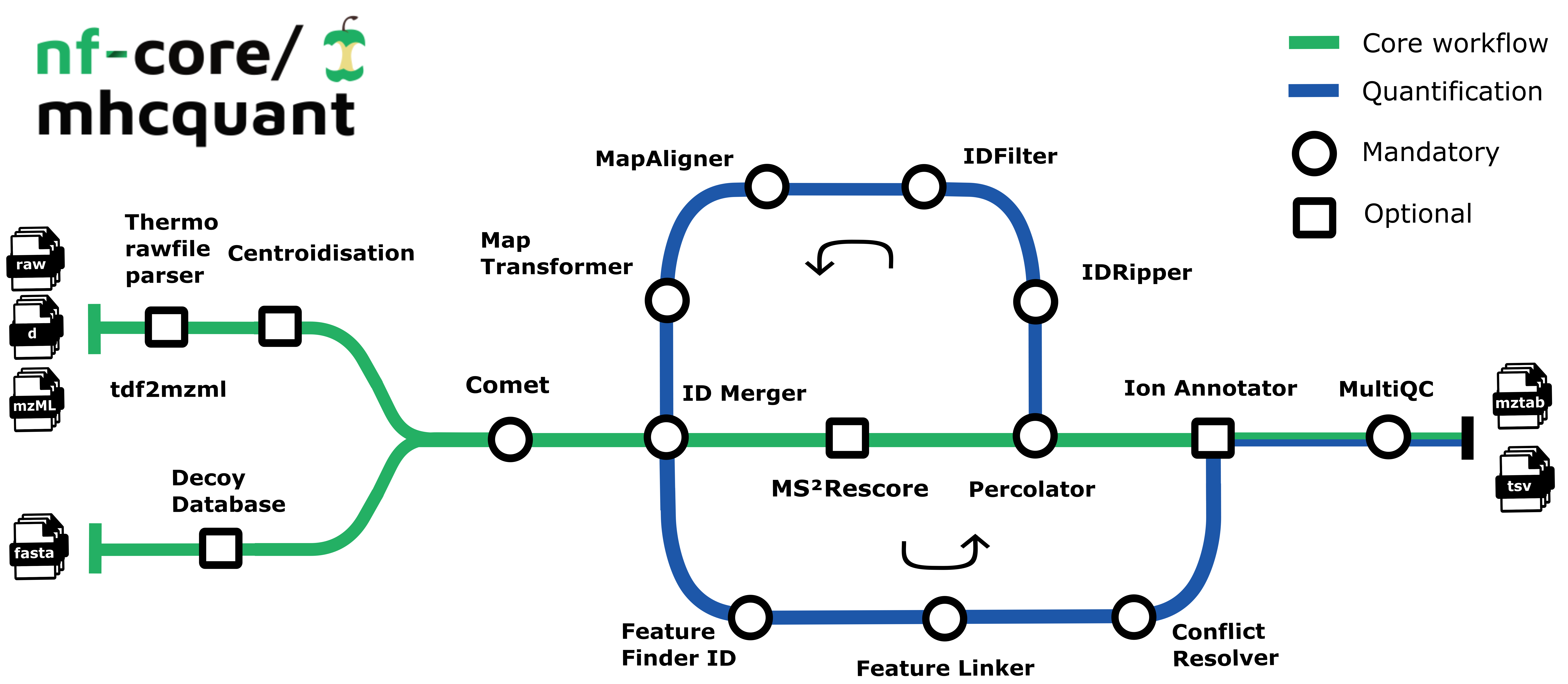
The pipeline is based on the OpenMS C++ framework for computational mass spectrometry. Spectrum files (mzML/Thermo raw/Bruker tdf) serve as inputs and a database search (Comet) is performed based on a given input protein database. Peptide properties are predicted by MS²Rescore. FDR rescoring is applied using Percolator or Mokapot based on a competitive target-decoy approach. The pipeline supports both local FDR control (per sample-condition group) and global FDR control (across all samples). For label-free quantification, all input files undergo identification-based retention time alignment and targeted feature extraction matching ids between runs. The pipeline can also generate spectrum libraries suitable for DIA-based searches as well as computing consensus epitopes using epicore.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where possible, these processes have been submitted to and installed from nf-core/modules in order to make them available to all nf-core pipelines, and to everyone within the Nextflow community!
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Usage
If you are new to Nextflow and nf-core, please refer to this page on how
to set-up Nextflow. Make sure to test your setup
with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.tsv
ID Sample Condition ReplicateFileName
1 tumor treated /path/to/msrun1.raw|mzML|d
2 tumor treated /path/to/msrun2.raw|mzML|d
3 tumor untreated /path/to/msrun3.raw|mzML|d
4 tumor untreated /path/to/msrun4.raw|mzML|dEach row represents a mass spectrometry run in one of the formats: raw, RAW, mzML, mzML.gz, d, d.tar.gz, d.zip
Now, you can run the pipeline using:
nextflow run nf-core/mhcquant \
-profile <docker/singularity/.../institute> \
--input 'samplesheet.tsv' \
--fasta 'SWISSPROT_2020.fasta' \
--outdir ./resultsOptional parameters for additional functionality:
# Enable quantification, global FDR and spectrum library generation, ion annotations, and consenus epitopes
nextflow run nf-core/mhcquant \
--input 'samplesheet.tsv' \
--fasta 'SWISSPROT_2020.fasta' \
--annotate_ions \
--epicore \
--generate_speclib \
--global_fdr \
--quantify \
--outdir ./results \
-profile dockerPlease provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline summary
Default Steps
By default the pipeline currently performs identification of MHC class I peptides with HCD settings:
- Spectra Preparation: Preparing spectra dependent on the input format (
PREPARE_SPECTRAsubworkflow) - Database Preparation: Creation of reversed decoy database (
DecoyDatabase) - Peptide Identification: Identification of peptides in the MS/MS spectra (
CometAdapter) - Database Indexing: Refreshes protein references for all peptide hits and adds target/decoy information (
PeptideIndexer) - Identification Merging: Merges identification files with the same
SampleandConditionlabel (IDMerger) - Rescoring: Feature prediction and peptide-spectrum-match rescoring (
RESCOREsubworkflow)- Prediction of retention times and MS2 intensities (
MS²Rescore) - Extract PSM features for rescoring engines (
PSMFeatureExtractor) - Peptide-spectrum-match rescoring using Percolator or Mokapot (
PercolatorAdapter) - Filters peptide identification result according to configurable FDR threshold (
IDFilter)
- Prediction of retention times and MS2 intensities (
- Export: Converts identification result to tab-separated files (
TextExporter)
FDR Control Modes
The pipeline supports two FDR control strategies:
- Local FDR (default): FDR control applied per
SampleandConditiongroup - Global FDR: FDR control applied across all samples in the dataset (enable with
--global_fdr)
Additional Steps
Additional functionality contained by the pipeline currently includes:
Quantification (QUANT subworkflow)
When enabled with --quantify, the pipeline performs label-free quantification:
- Alignment: Corrects retention time distortions between runs (
MAP_ALIGNMENTsubworkflow)- Corrects retention time distortions between runs (
MapAlignerIdentification) - Applies retention time transformations to runs (
MapRTTransformer)
- Corrects retention time distortions between runs (
- Feature Processing: Detects and processes features (
PROCESS_FEATUREsubworkflow)- Detects features in MS1 data based on peptide identifications (
FeatureFinderIdentification) - Group corresponding features across label-free experiments (
FeatureLinkerUnlabeledKD) - Resolves ambiguous annotations of features with peptide identifications (
IDConflictResolver)
- Detects features in MS1 data based on peptide identifications (
Spectrum Library Generation (SPECLIB subworkflow)
When enabled with --generate_speclib, the pipeline generates spectrum libraries suitable for DIA-based searches. Outputs one library per sample or a single library across all samples (if global FDR mode is enabled with --global_fdr).
Ion Annotation (IONANNOTATOR subworkflow)
The pipeline annotates the final list of peptides with their respective ions and charges:
- Annotates final list of peptides with their respective ions and charges (
IonAnnotator)
Output
Documentation
To see the the results of a test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
- Nextflow installation
- Pipeline configuration
- Running the pipeline
- This includes tutorials, FAQs, and troubleshooting instructions
- Output and how to interpret the results
Credits
nf-core/mhcquant was originally written by Leon Bichmann from the Kohlbacher Lab. The pipeline was re-written in Nextflow DSL2 by Marissa Dubbelaar and was significantly improved by Jonas Scheid and Steffen Lemke from Peptide-based Immunotherapy and Quantitative Biology Center in Tübingen.
Helpful contributors:
- Lukas Heumos
- Alexander Peltzer
- Maxime Garcia
- Gisela Gabernet
- Susanne Jodoin
- Oskar Wacker
- Leon Kuchenbecker
- Phil Ewels
- Christian Fufezan
- Sven Fillinger
- Kevin Menden
- Julia Graf
- Jana Hoffmann
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #mhcquant channel (you can join with this invite).
Citations
If you use nf-core/mhcquant for your analysis, please cite the corresponding manuscript: 10.1186/s13059-025-03763-8
MHCquant2 refines immunopeptidomics tumor antigen discovery
Jonas Scheid, Steffen Lemke, Naomi Hoenisch-Gravel, Anna Dengler, Timo Sachsenberg, Arthur Declerq, Ralf Gabriels, Jens Bauer, Marcel Wacker, Leon Bichmann, Lennart Martens, Marissa L. Dubbelaar, Sven Nahnsen & Juliane S. Walz
Genome Biology 2025 26 (1), 290. doi: 10.1021/acs.jproteome.9b00313
MHCquant: Automated and Reproducible Data Analysis for Immunopeptidomics
Leon Bichmann, Annika Nelde, Michael Ghosh, Lukas Heumos, Christopher Mohr, Alexander Peltzer, Leon Kuchenbecker, Timo Sachsenberg, Juliane S. Walz, Stefan Stevanović, Hans-Georg Rammensee & Oliver Kohlbacher
Journal of Proteome Research 2019 18 (11), 3876-3884. doi: 10.1021/acs.jproteome.9b00313
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.
In addition, references of tools and data used in this pipeline are as follows:
OpenMS framework
Pfeuffer J. et al, Nat Methods 2024 Mar;21(3):365-367. doi: 0.1038/s41592-024-02197-7.
Comet Search Engine
Eng J.K. et al, J Am Soc Mass Spectrom. 2015 Nov;26(11):1865-74. doi: 10.1007/s13361-015-1179-x.
Retention time prediction
Bouwmeester R. et al, Nature Methods 2021 Oct;18(11):1363-1369. doi: 10.1038/s41592-021-01301-5
MS² Peak intensity prediction
Declercq A. et al, Nucleic Acids Res. 2023 Jul 5;51(W1):W338-W342. doi: 10.1093/nar/gkad335
CCS prediction
Declercq A. et al Journal of Proteome Research 2025 Feb 6. doi: 10.1021/acs.jproteome.4c00609
MS²Rescore framework
Buur L. M. et al, _J Proteome Res. 2024 Mar 16. doi: 10.1021/acs.jproteome.3c00785
Percolator
Käll L. et al, Nat Methods 2007 Nov;4(11):923-5. doi: 10.1038/nmeth1113.
Identification based RT Alignment
Weisser H. et al, J Proteome Res. 2013 Apr 5;12(4):1628-44. doi: 10.1021/pr300992u
Targeted peptide quantification
Weisser H. et al, J Proteome Res. 2017 Aug 4;16(8):2964-2974. doi: 10.1021/acs.jproteome.7b00248