nf-core/scrnaseq
Single-cell RNA-Seq pipeline for barcode-based protocols such as 10x, DropSeq or SmartSeq, offering a variety of aligners and empty-droplet detection
2.5.0). The latest
stable release is
4.1.0
.
Introduction
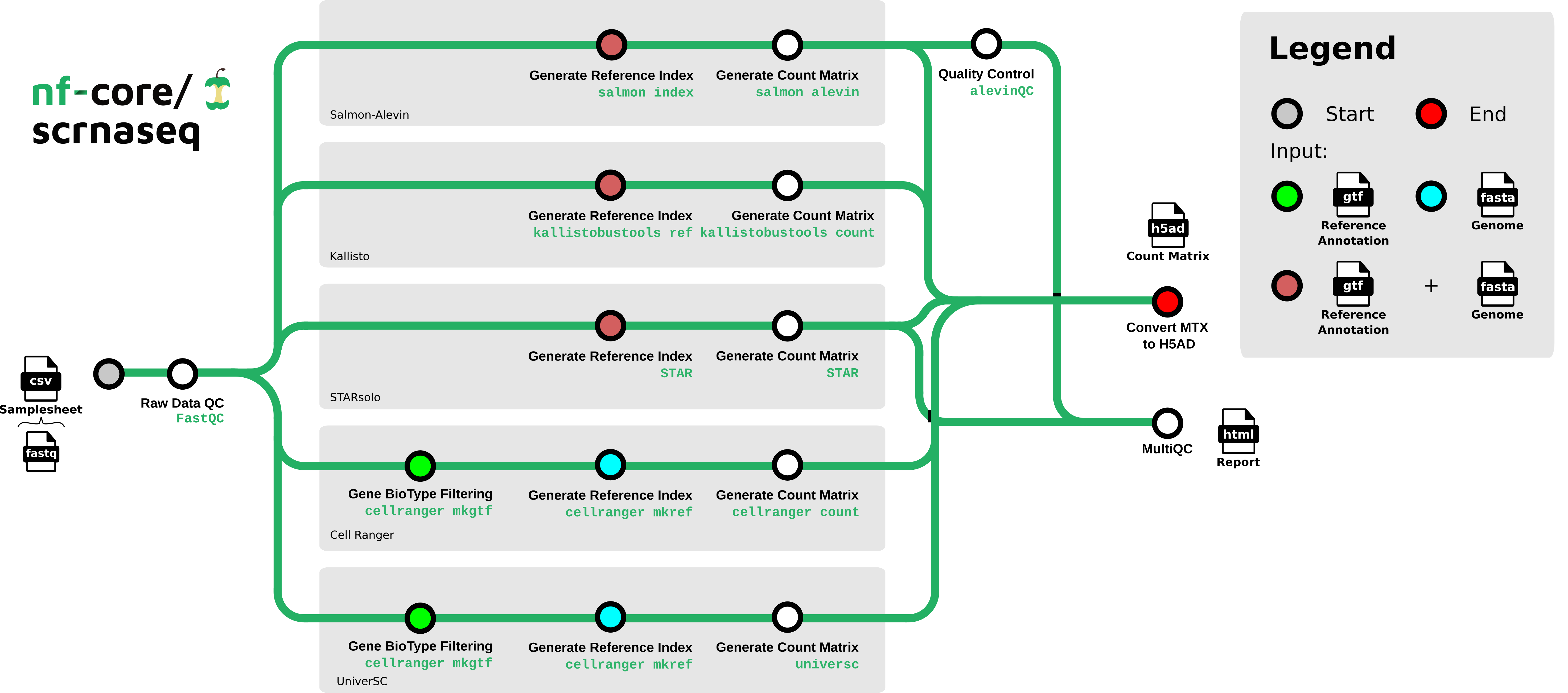
nf-core/scrnaseq is a bioinformatics best-practice analysis pipeline for processing 10x Genomics single-cell RNA-seq data.
This is a community effort in building a pipeline capable to support:
- Alevin-Fry + AlevinQC
- STARSolo
- Kallisto + BUStools
- Cellranger
- UniverSC
Documentation
The nf-core/scrnaseq pipeline comes with documentation about the pipeline usage, parameters and output.
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,fastq_1,fastq_2,expected_cells
pbmc8k,pbmc8k_S1_L007_R1_001.fastq.gz,pbmc8k_S1_L007_R2_001.fastq.gz,"10000"
pbmc8k,pbmc8k_S1_L008_R1_001.fastq.gz,pbmc8k_S1_L008_R2_001.fastq.gz,"10000"Each row represents a fastq file (single-end) or a pair of fastq files (paired end).
Now, you can run the pipeline using:
nextflow run nf-core/scrnaseq \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--genome_fasta GRCm38.p6.genome.chr19.fa \
--gtf gencode.vM19.annotation.chr19.gtf \
--protocol 10XV2 \
--aligner <alevin/kallisto/star/cellranger/universc> \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Decision Tree for users
The nf-core/scrnaseq pipeline features several paths to analyze your single cell data. Future additions will also be done soon, e.g. the addition of multi-ome analysis types. To aid users in analyzing their data, we have added a decision tree to help people decide on what type of analysis they want to run and how to choose appropriate parameters for that.
Loading graph
Options for the respective alignment method can be found here to choose between methods.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/scrnaseq was originally written by Bailey PJ, Botvinnik O, Marques de Almeida F, Gabernet G, Peltzer A, Sturm G.
We thank the following people and teams for their extensive assistance in the development of this pipeline:
- @heylf
- @KevinMenden
- @FloWuenne
- @rob-p
- GHGA
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #scrnaseq channel (you can join with this invite).
Citations
If you use nf-core/scrnaseq for your analysis, please cite it using the following doi: 10.5281/zenodo.3568187
The basic benchmarks that were used as motivation for incorporating the three available modular workflows can be found in this publication.
We offer all three paths for the processing of scRNAseq data so it remains up to the user to decide which pipeline workflow is chosen for a particular analysis question.
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.