nf-core/airrflow
B-cell and T-cell Adaptive Immune Receptor Repertoire (AIRR) sequencing analysis pipeline using the Immcantation framework
3.0). The latest
stable release is
5.0.0
.
Introduction
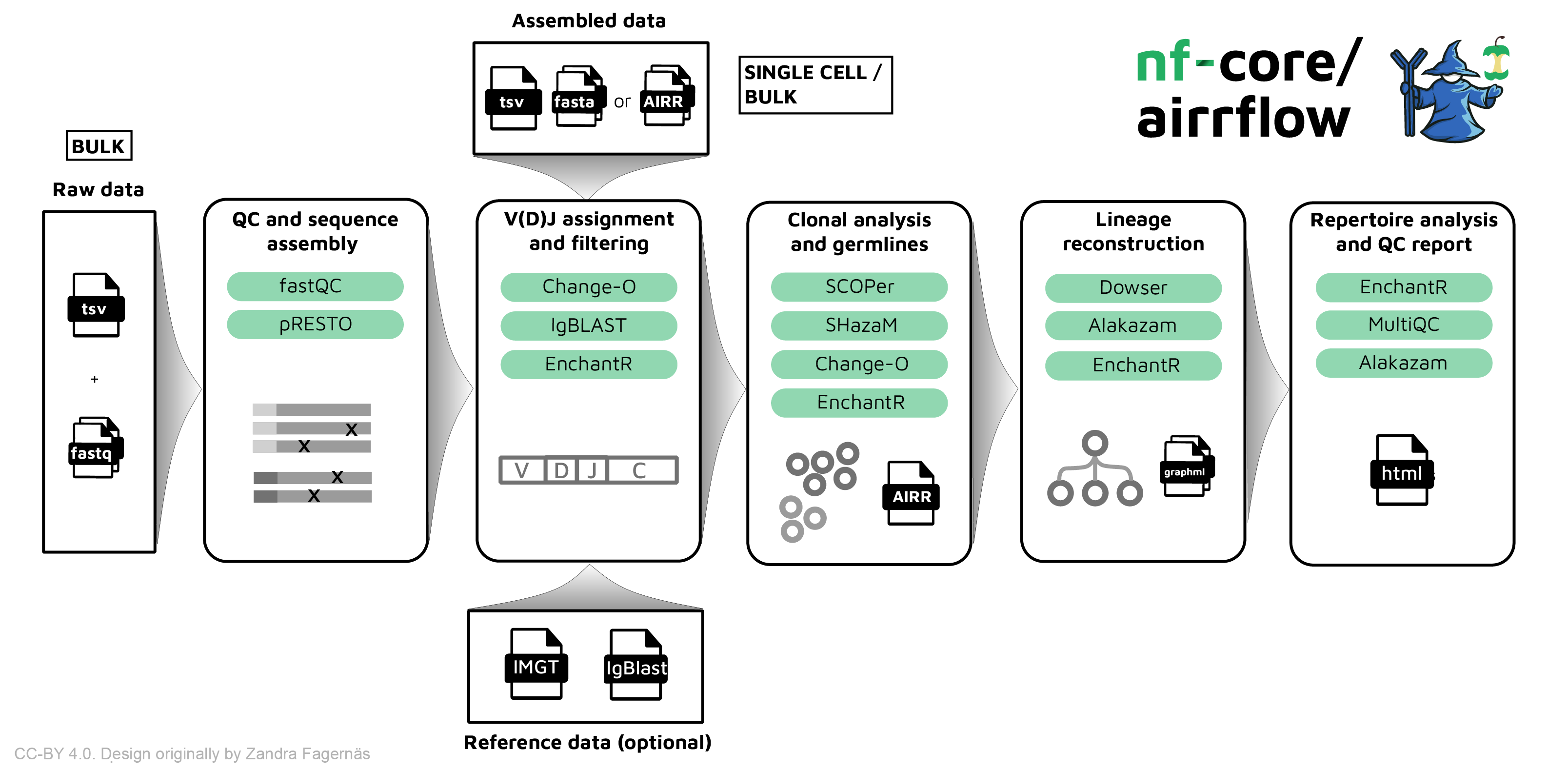
** nf-core/airrflow ** is a bioinformatics best-practice pipeline to analyze B-cell or T-cell repertoire sequencing data. It makes use of the Immcantation toolset. The input data can be targeted amplicon bulk sequencing data of the V, D, J and C regions of the B/T-cell receptor with multiplex PCR or 5’ RACE protocol, or assembled reads (bulk or single cell).
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where possible, these processes have been submitted to and installed from nf-core/modules in order to make them available to all nf-core pipelines, and to everyone within the Nextflow community!
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world datasets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Pipeline summary
nf-core/airrflow allows the end-to-end processing of BCR and TCR bulk and single cell targeted sequencing data. Several protocols are supported, please see the usage documenation for more details on the supported protocols.
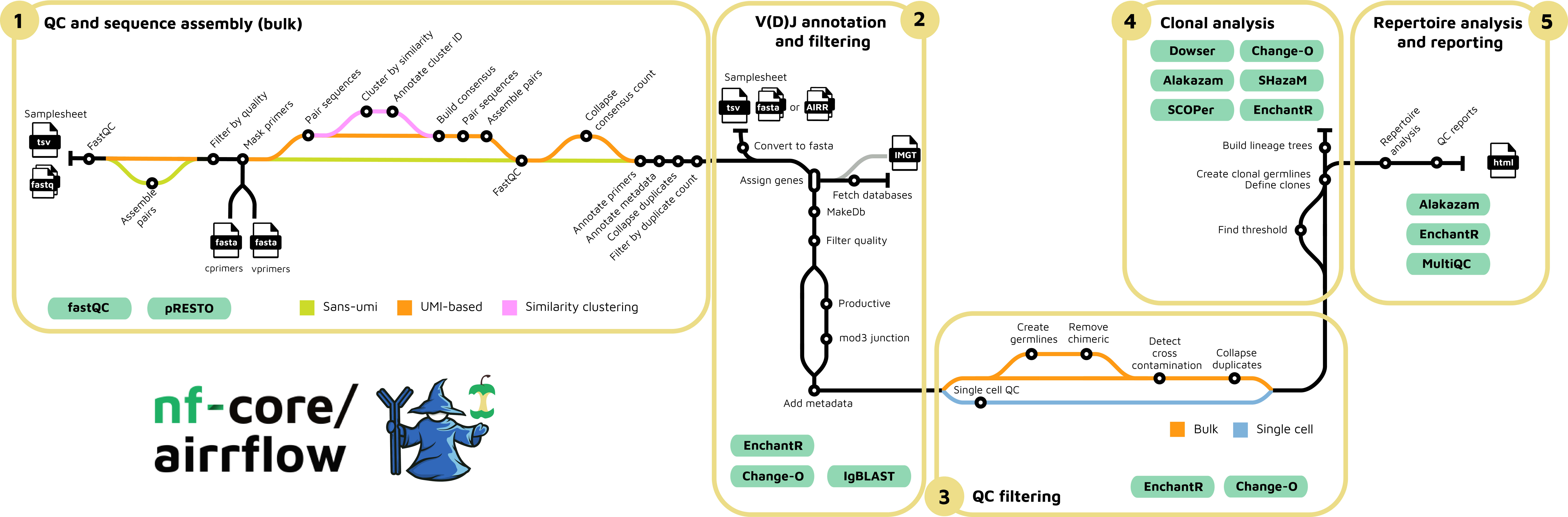
- QC and sequence assembly (bulk only)
- Raw read quality control, adapter trimming and clipping (
Fastp). - Filter sequences by base quality (
pRESTO FilterSeq). - Mask amplicon primers (
pRESTO MaskPrimers). - Pair read mates (
pRESTO PairSeq). - For UMI-based sequencing:
- Cluster sequences according to similarity (optional for insufficient UMI diversity) (
pRESTO ClusterSets). - Build consensus of sequences with the same UMI barcode (
pRESTO BuildConsensus).
- Cluster sequences according to similarity (optional for insufficient UMI diversity) (
- Assemble R1 and R2 read mates (
pRESTO AssemblePairs). - Remove and annotate read duplicates (
pRESTO CollapseSeq). - Filter out sequences that do not have at least 2 duplicates (
pRESTO SplitSeq).
- V(D)J annotation and filtering (bulk and single-cell)
- Assign gene segments with
IgBlastusing the IMGT database (Change-O AssignGenes). - Annotate alignments in AIRR format (
Change-O MakeDB) - Filter by alignment quality (locus matching v_call chain, min 200 informative positions, max 10% N nucleotides)
- Filter productive sequences (
Change-O ParseDB split) - Filter junction length multiple of 3
- Annotate metadata (
EnchantR)
- QC filtering (bulk and single-cell)
- Bulk sequencing filtering:
- Remove chimeric sequences (optional) (
SHazaM,EnchantR) - Detect cross-contamination (optional) (
EnchantR) - Collapse duplicates (
Alakazam,EnchantR)
- Remove chimeric sequences (optional) (
- Single-cell QC filtering (
EnchantR)- Remove cells without heavy chains.
- Remove cells with multiple heavy chains.
- Remove sequences in different samples that share the same
cell_idand nucleotide sequence. - Modify
cell_ids to ensure they are unique in the project.
- Clonal analysis (bulk and single-cell)
- Find threshold for clone definition (
SHazaM,EnchantR). - Create germlines and define clones, repertoire analysis (
Change-O,EnchantR). - Build lineage trees (
SCOPer,IgphyML,EnchantR).
- Repertoire analysis and reporting
- Custom repertoire analysis pipeline report (
Alakazam). - Aggregate QC reports (
MultiQC).
Quick Start
-
Install
Nextflow(>=22.10.1) -
Install any of
Docker,Singularity(you can follow this tutorial),Podman,ShifterorCharliecloudfor full pipeline reproducibility (you can useCondaboth to install Nextflow itself and also to manage software within pipelines. Please only use it within pipelines as a last resort; see docs). -
Download the pipeline and test it on a minimal dataset with a single command:
nextflow run nf-core/airrflow -profile test,<docker/singularity/podman/shifter/charliecloud/conda/institute> --outdir <OUTDIR>Note that some form of configuration will be needed so that Nextflow knows how to fetch the required software. This is usually done in the form of a config profile (
YOURPROFILEin the example command above). You can chain multiple config profiles in a comma-separated string.- The pipeline comes with config profiles called
docker,singularity,podman,shifter,charliecloudandcondawhich instruct the pipeline to use the named tool for software management. For example,-profile test,docker. - Please check nf-core/configs to see if a custom config file to run nf-core pipelines already exists for your Institute. If so, you can simply use
-profile <institute>in your command. This will enable eitherdockerorsingularityand set the appropriate execution settings for your local compute environment. - If you are using
singularity, please use thenf-core downloadcommand to download images first, before running the pipeline. Setting theNXF_SINGULARITY_CACHEDIRorsingularity.cacheDirNextflow options enables you to store and re-use the images from a central location for future pipeline runs. - If you are using
conda, it is highly recommended to use theNXF_CONDA_CACHEDIRorconda.cacheDirsettings to store the environments in a central location for future pipeline runs.
- The pipeline comes with config profiles called
-
Start running your own analysis!
nextflow run nf-core/airrflow \
-profile <docker/singularity/podman/shifter/charliecloud/conda/institute> \
--input samplesheet.tsv \
--library_generation_method specific_pcr_umi \
--cprimers CPrimers.fasta \
--vprimers VPrimers.fasta \
--umi_length 12 \
--max_memory 8.GB \
--max_cpus 8 \
--outdir ./resultsSee usage docs for all of the available options when running the pipeline.
Documentation
The nf-core/airrflow pipeline comes with documentation about the pipeline usage, parameters and output.
Credits
nf-core/airrflow was written by Gisela Gabenet, Susanna Marquez, Alexander Peltzer and Simon Heumos.
Further contributors to the pipeline are:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #airrflow channel (you can join with this invite).
Citations
If you use nf-core/airrflow for your analysis, please cite it using the following DOI: 10.5281/zenodo.2642009
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.