nf-core/rnaseq
RNA sequencing analysis pipeline using STAR, RSEM, HISAT2 or Salmon with gene/isoform counts and extensive quality control.
1.4.2). The latest stable release is3.26.0.Output
This document describes the output produced by the pipeline. Most of the plots are taken from the MultiQC report, which summarises results at the end of the pipeline.
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
- FastQC - read quality control
- TrimGalore - adapter trimming
- SortMeRNA - ribosomal RNA removal
- STAR - alignment
- RSeQC - RNA quality control metrics
- Qualimap - RNA quality control metrics
- dupRadar - technical / biological read duplication
- Preseq - library complexity
- featureCounts - gene counts, biotype counts, rRNA estimation.
- Salmon - gene counts, transcripts counts.
- tximport - gene counts, transcripts counts, SummarizedExperimment object.
- StringTie - FPKMs for genes and transcripts
- Sample_correlation - create MDS plot and sample pairwise distance heatmap / dendrogram
- MultiQC - aggregate report, describing results of the whole pipeline
FastQC
FastQC gives general quality metrics about your reads. It provides information about the quality score distribution across your reads, the per base sequence content (%T/A/G/C). You get information about adapter contamination and other overrepresented sequences.
For further reading and documentation see the FastQC help.
NB: The FastQC plots displayed in the MultiQC report shows untrimmed reads. They may contain adapter sequence and potentially regions with low quality. To see how your reads look after trimming, look at the FastQC reports in the
trim_galoredirectory.
Output directory: results/fastqc
sample_fastqc.html- FastQC report, containing quality metrics for your untrimmed raw fastq files
zips/sample_fastqc.zip- zip file containing the FastQC report, tab-delimited data file and plot images
TrimGalore
The nfcore/rnaseq pipeline uses TrimGalore for removal of adapter contamination and trimming of low quality regions. TrimGalore uses Cutadapt for adapter trimming and runs FastQC after it finishes.
MultiQC reports the percentage of bases removed by TrimGalore in the General Statistics table, along with a line plot showing where reads were trimmed.
Output directory: results/trim_galore
Contains FastQ files with quality and adapter trimmed reads for each sample, along with a log file describing the trimming.
sample_val_1.fq.gz,sample_val_2.fq.gz- Trimmed FastQ data, reads 1 and 2.
- NB: Only saved if
--saveTrimmedhas been specified.
logs/sample_val_1.fq.gz_trimming_report.txt- Trimming report (describes which parameters that were used)
FastQC/sample_val_1_fastqc.zip- FastQC report for trimmed reads
Single-end data will have slightly different file names and only one FastQ file per sample.
SortMeRNA
When --removeRiboRNA is specified, nfcore/rnaseq pipeline uses SortMeRNA for removal of rRNA. SortMeRNA requires reference sequences and these are by default from the SILVA database.
Output directory: results/SortMeRNA
Contains FastQ files with quality and adapter trimmed reads for each sample, along with a log file describing the trimming.
reads/sample-fw.fq.gz,reads/sample-rv.fq.gz- Trimmed and rRNA depleted FastQ data, reads forward and reverse.
- NB: Only saved if
--save_nonrRNA_readshas been specified.
logs/sample_rRNA_report.txt- Report how many reads where removed due to matches to reference database(s).
Single-end data will have slightly different file names (reads/sample.fq.gz) and only one FastQ file per sample.
STAR
STAR is a read aligner designed for RNA sequencing. STAR stands for Spliced Transcripts Alignment to a Reference, it produces results comparable to TopHat (the aligned previously used by NGI for RNA alignments) but is much faster.
The STAR section of the MultiQC report shows a bar plot with alignment rates: good samples should have most reads as Uniquely mapped and few Unmapped reads.
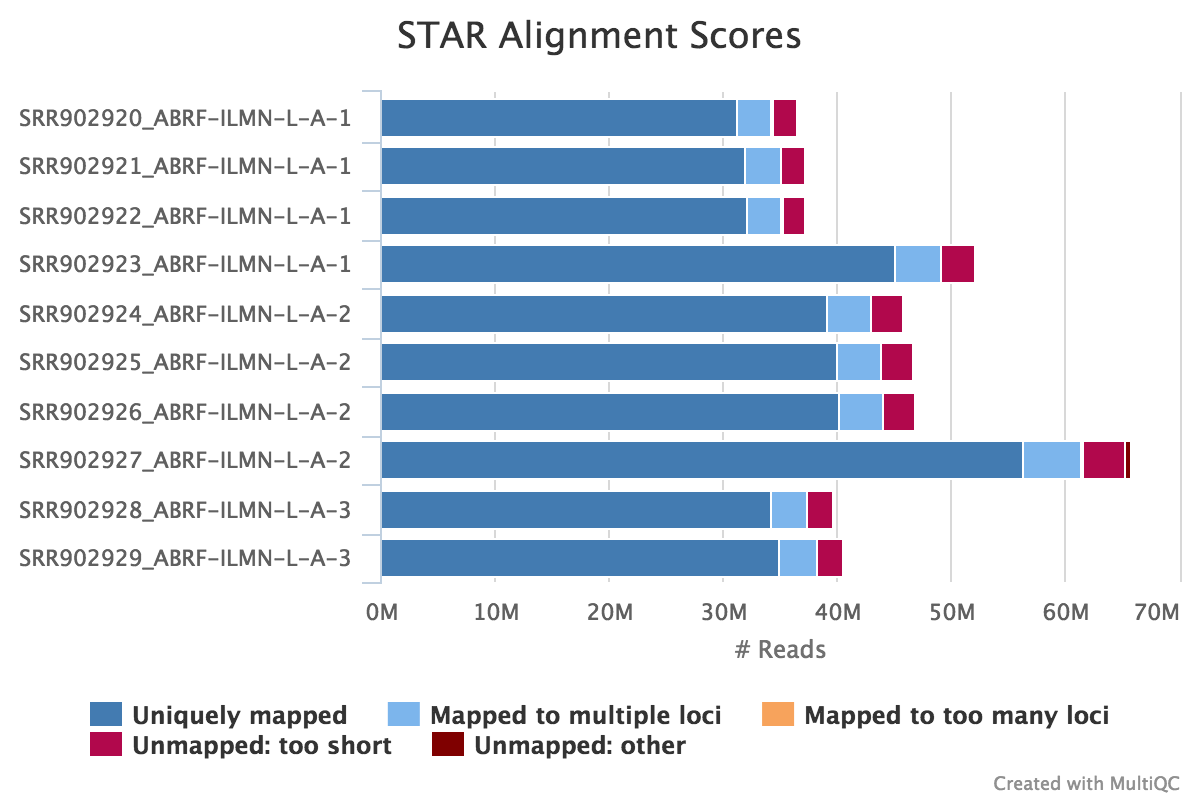
Output directory: results/STAR
Sample_Aligned.sortedByCoord.out.bam- The aligned BAM file
Sample_Log.final.out- The STAR alignment report, contains mapping results summary
Sample_Log.outandSample_Log.progress.out- STAR log files, containing a lot of detailed information about the run. Typically only useful for debugging purposes.
Sample_SJ.out.tab- Filtered splice junctions detected in the mapping
unaligned/...- Contains the unmapped reads that couldn’t be mapped against the reference genome chosen. This is only available when the user specifically asks for
--saveUnalignedoutput.
- Contains the unmapped reads that couldn’t be mapped against the reference genome chosen. This is only available when the user specifically asks for
RSeQC
RSeQC is a package of scripts designed to evaluate the quality of RNA seq data. You can find out more about the package at the RSeQC website.
This pipeline runs several, but not all RSeQC scripts. All of these results are summarised within the MultiQC report and described below.
Output directory: results/rseqc
These are all quality metrics files and contains the raw data used for the plots in the MultiQC report. In general, the .r files are R scripts for generating the figures, the .txt are summary files, the .xls are data tables and the .pdf files are summary figures.
BAM stat
Output: Sample_bam_stat.txt
This script gives numerous statistics about the aligned BAM files produced by STAR. A typical output looks as follows:
#Output (all numbers are read count)#==================================================Total records: 41465027QC failed: 0Optical/PCR duplicate: 0Non Primary Hits 8720455Unmapped reads: 0
mapq < mapq_cut (non-unique): 3127757mapq >= mapq_cut (unique): 29616815Read-1: 14841738Read-2: 14775077Reads map to '+': 14805391Reads map to '-': 14811424Non-splice reads: 25455360Splice reads: 4161455Reads mapped in proper pairs: 21856264Proper-paired reads map to different chrom: 7648MultiQC plots each of these statistics in a dot plot. Each sample in the project is a dot - hover to see the sample highlighted across all fields.
RSeQC documentation: bam_stat.py
Infer experiment
Output: Sample_infer_experiment.txt
This script predicts the mode of library preparation (sense-stranded or antisense-stranded) according to how aligned reads overlay gene features in the reference genome. Example output from an unstranded (~50% sense/antisense) library of paired end data:
From MultiQC report:
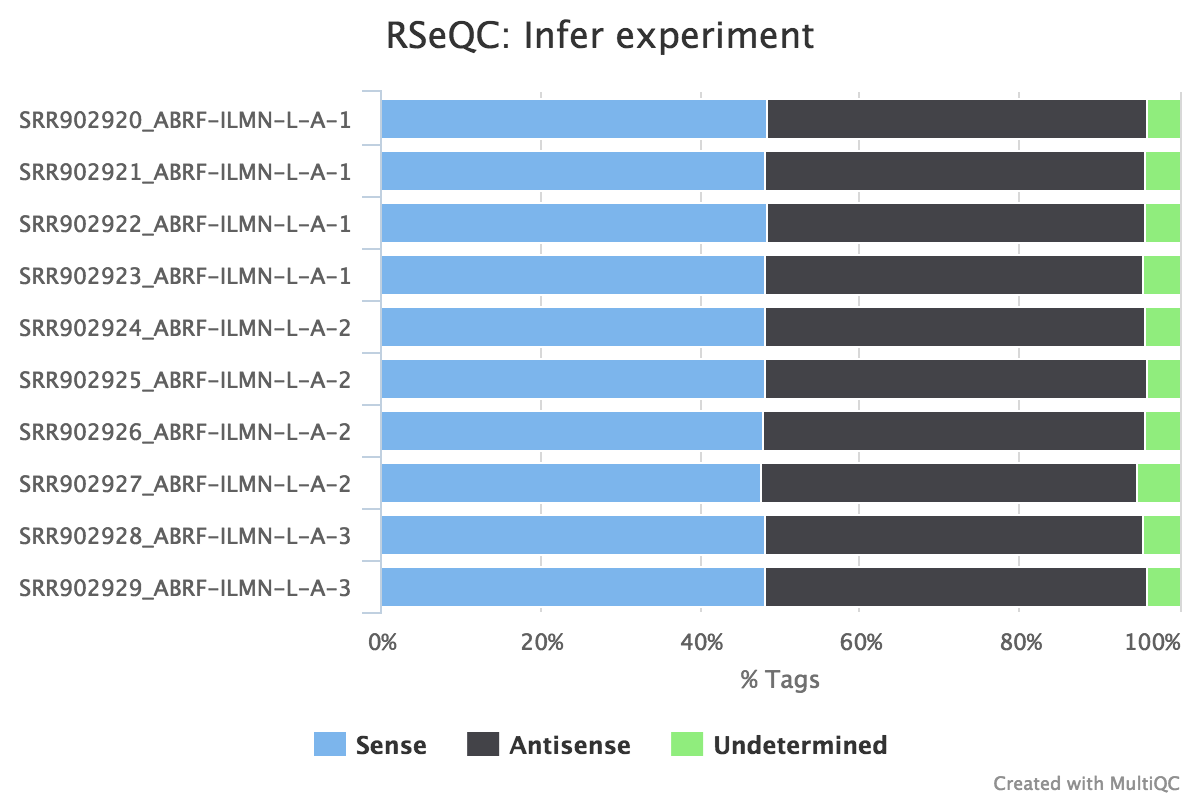
From the infer_experiment.txt file:
This is PairEnd DataFraction of reads failed to determine: 0.0409Fraction of reads explained by "1++,1--,2+-,2-+": 0.4839Fraction of reads explained by "1+-,1-+,2++,2--": 0.4752RSeQC documentation: infer_experiment.py
Junction saturation
Output:
Sample_rseqc.junctionSaturation_plot.pdfSample_rseqc.junctionSaturation_plot.r
This script shows the number of splice sites detected at the data at various levels of subsampling. A sample that reaches a plateau before getting to 100% data indicates that all junctions in the library have been detected, and that further sequencing will not yield more observations. A good sample should approach such a plateau of Known junctions, very deep sequencing is typically requires to saturate all Novel Junctions in a sample.
None of the lines in this example have plateaued and thus these samples could reveal more alternative splicing information if they were sequenced deeper.
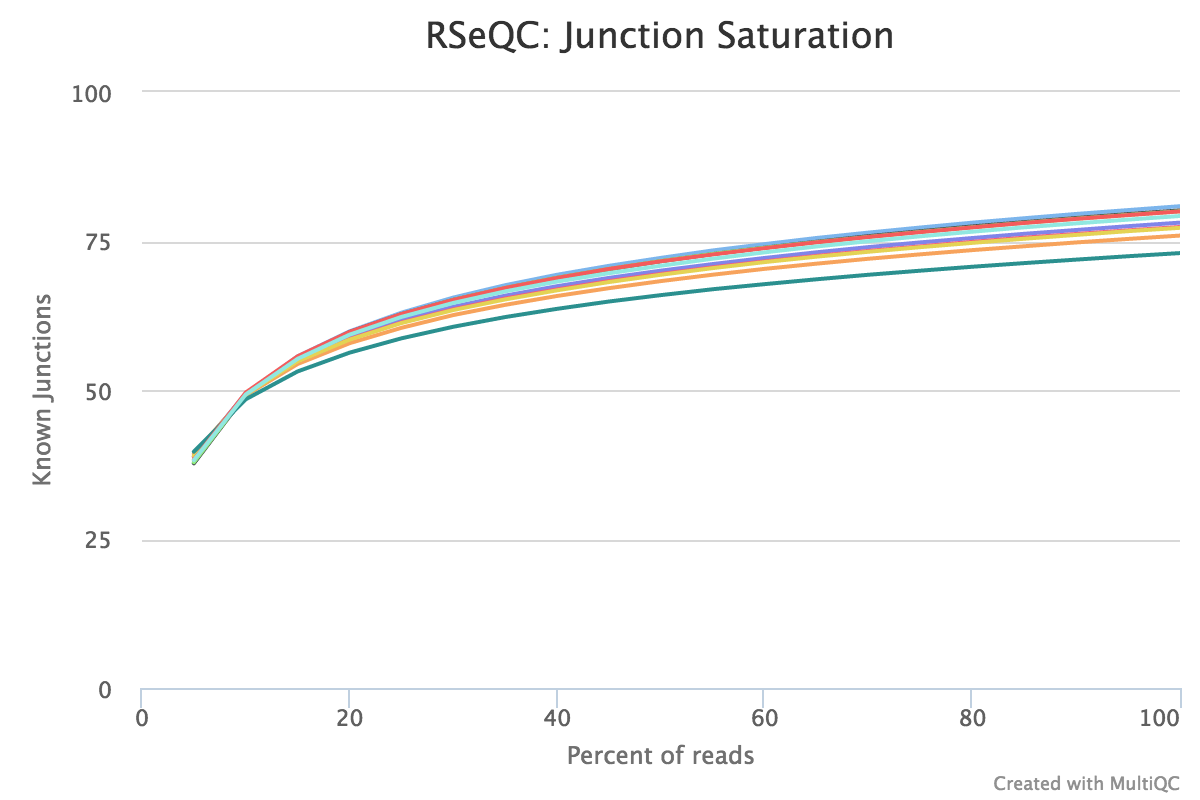
RSeQC documentation: junction_saturation.py
RPKM saturation
Output:
Sample_RPKM_saturation.eRPKM.xlsSample_RPKM_saturation.rawCount.xlsSample_RPKM_saturation.saturation.pdfSample_RPKM_saturation.saturation.r
This tool resamples a subset of the total RNA reads and calculates the RPKM value for each subset. We use the default subsets of every 5% of the total reads. A percent relative error is then calculated based on the subsamples; this is the y-axis in the graph. A typical PDF figure looks as follows:
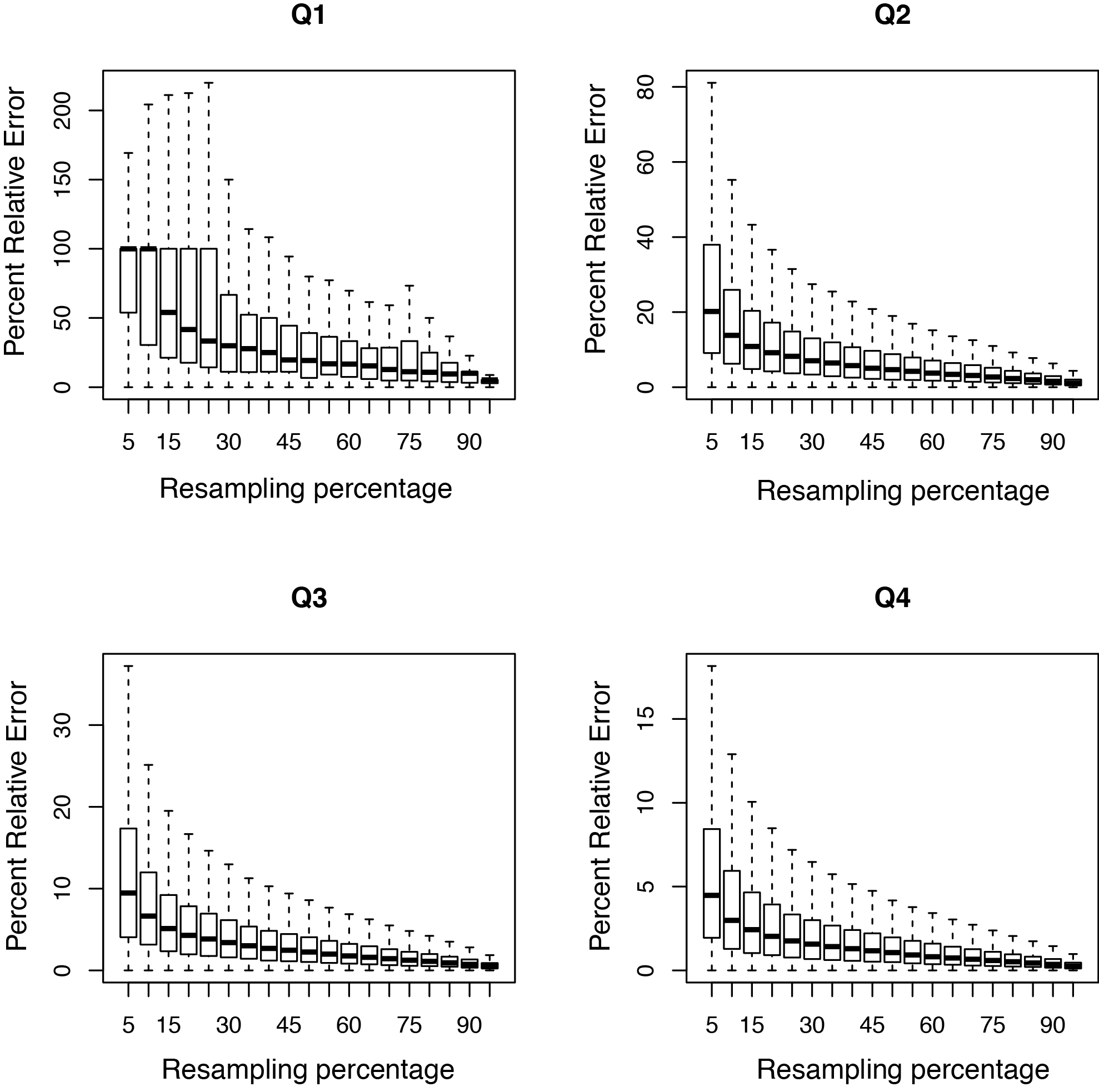
A complex library will have low resampling error in well expressed genes.
This data is not currently reported in the MultiQC report.
RSeQC documentation: RPKM_saturation.py
Read duplication
Output:
Sample_read_duplication.DupRate_plot.pdfSample_read_duplication.DupRate_plot.rSample_read_duplication.pos.DupRate.xlsSample_read_duplication.seq.DupRate.xls
This plot shows the number of reads (y-axis) with a given number of exact duplicates (x-axis). Most reads in an RNA-seq library should have a low number of exact duplicates. Samples which have many reads with many duplicates (a large area under the curve) may be suffering excessive technical duplication.
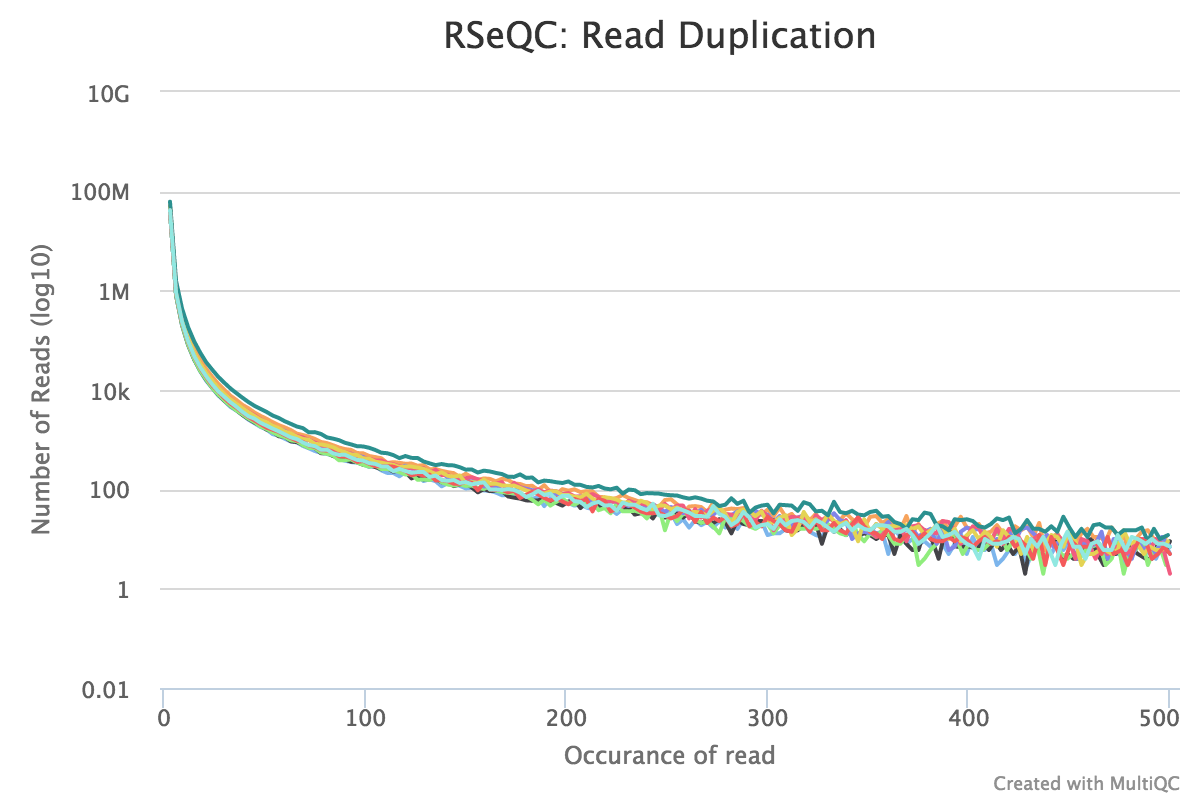
RSeQC documentation: read_duplication.py
Inner distance
Output:
Sample_rseqc.inner_distance.txtSample_rseqc.inner_distance_freq.txtSample_rseqc.inner_distance_plot.r
The inner distance script tries to calculate the inner distance between two paired RNA reads. It is the distance between the end of read 1 to the start of read 2,
and it is sometimes confused with the insert size (see this blog post for disambiguation):

Credit: modified from RSeQC documentation.
Note that values can be negative if the reads overlap. A typical set of samples may look like this:
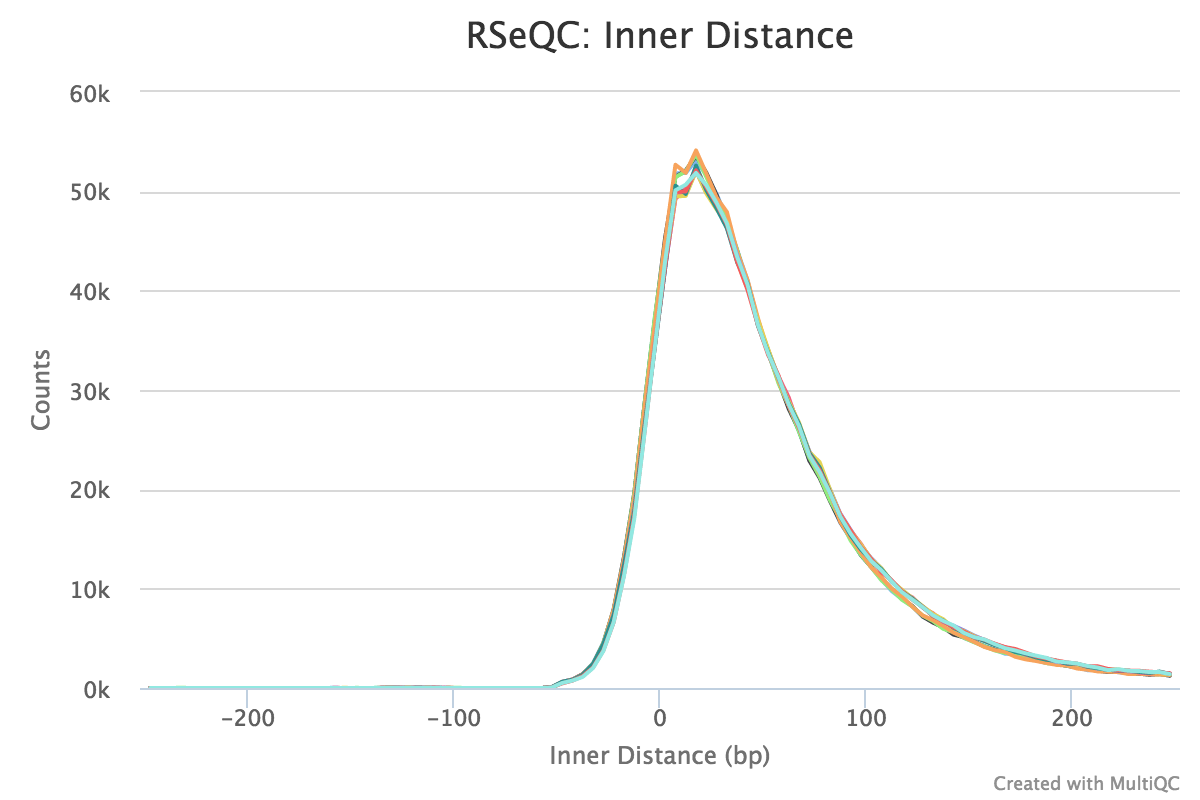
This plot will not be generated for single-end data. Very short inner distances are often seen in old or degraded samples (eg. FFPE).
RSeQC documentation: inner_distance.py
Read distribution
Output: Sample_read_distribution.txt
This tool calculates how mapped reads are distributed over genomic features. A good result for a standard RNA seq experiments is generally to have as many exonic reads as possible (CDS_Exons). A large amount of intronic reads could be indicative of DNA contamination in your sample or some other problem.
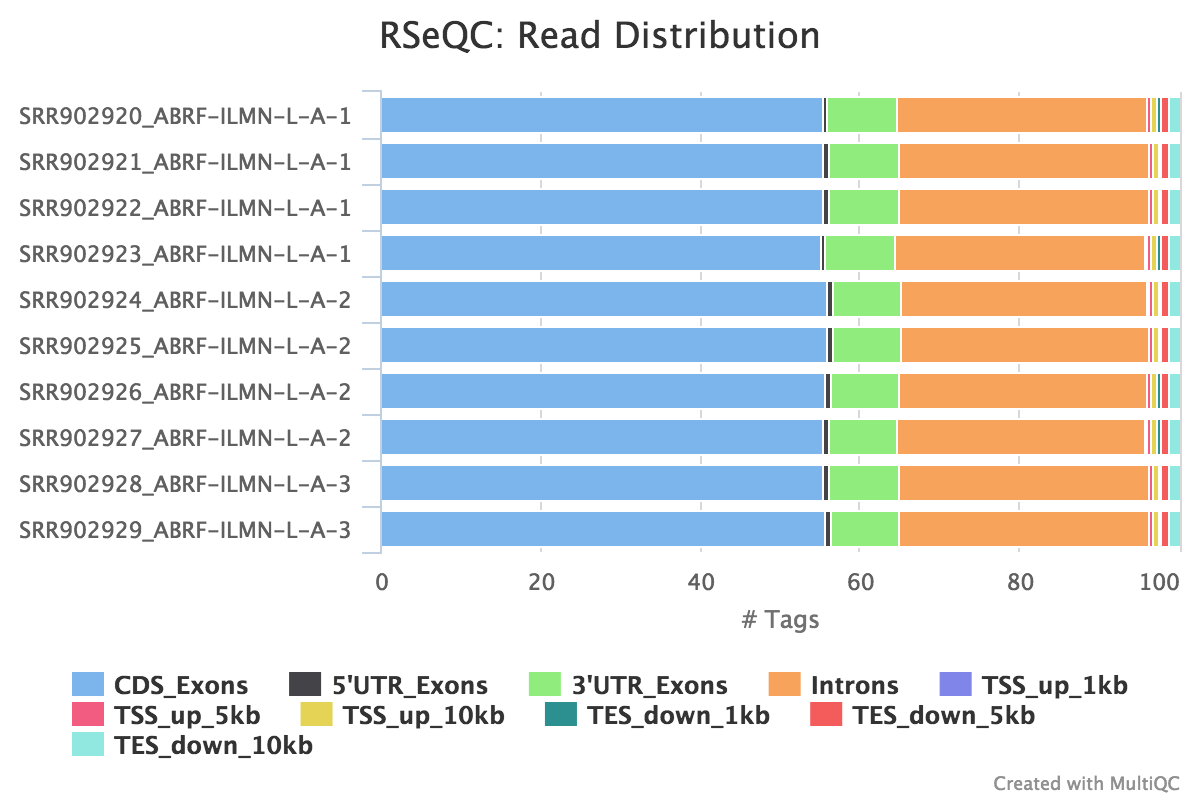
RSeQC documentation: read_distribution.py
Junction annotation
Output:
Sample_junction_annotation_log.txtSample_rseqc.junction.xlsSample_rseqc.junction_plot.rSample_rseqc.splice_events.pdfSample_rseqc.splice_junction.pdf
Junction annotation compares detected splice junctions to a reference gene model. An RNA read can be spliced 2 or more times, each time is called a splicing event.
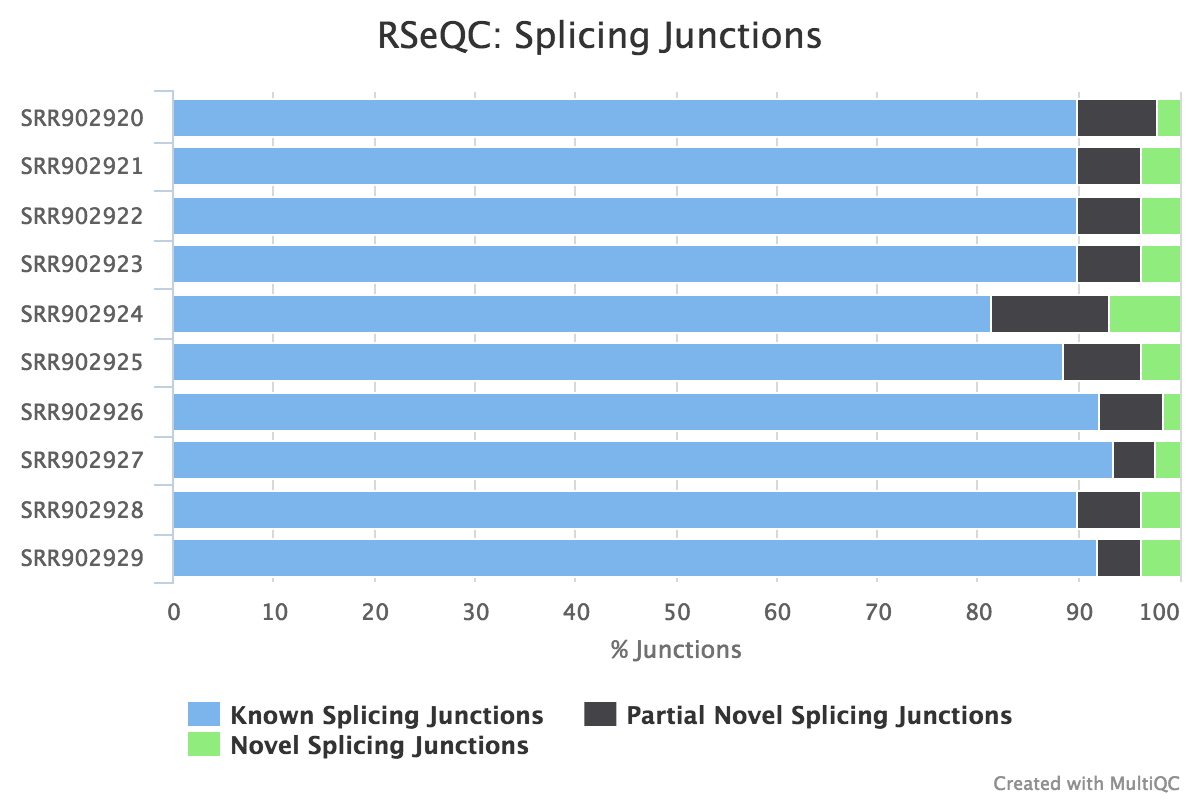
RSeQC documentation: junction_annotation.py
Qualimap
Qualimap is a standalone package written in java. It calculates read alignment assignment, transcript coverage, read genomic origin, junction analysis and 3’-5’ bias.
Output directory: results/qualimap
rnaseq_qc_results.txtqualimapReport.htmlcssraw_data_qualimapReportimages_qualimapReport
Qualimap RNAseq documentation: Qualimap docs.
dupRadar
dupRadar is a Bioconductor library for R. It plots the duplication rate against expression (RPKM) for every gene. A good sample with little technical duplication will only show high numbers of duplicates for highly expressed genes. Samples with technical duplication will have high duplication for all genes, irrespective of transcription level.
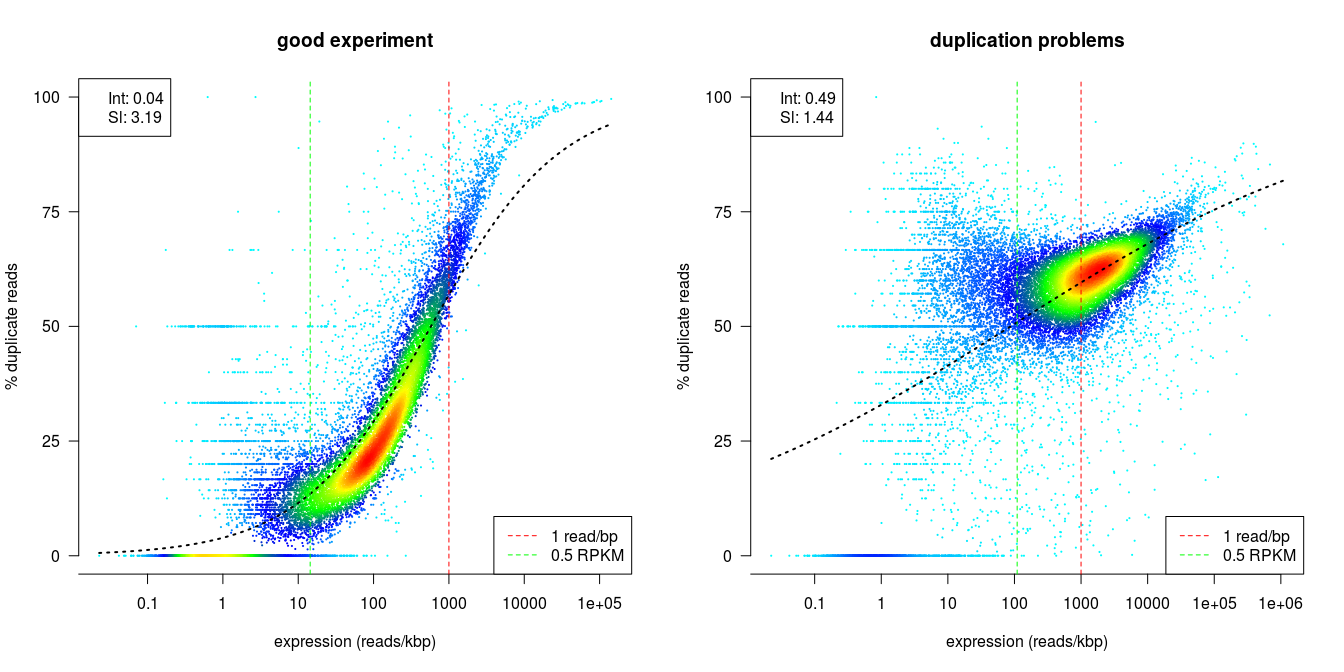
Credit: dupRadar documentation
Output directory: results/dupRadar
Sample_markDups.bam_duprateExpDens.pdfSample_markDups.bam_duprateExpBoxplot.pdfSample_markDups.bam_expressionHist.pdfSample_markDups.bam_dupMatrix.txtSample_markDups.bam_duprateExpDensCurve.txtSample_markDups.bam_intercept_slope.txt
DupRadar documentation: dupRadar docs
Preseq
Preseq estimates the complexity of a library, showing how many additional unique reads are sequenced for increasing the total read count. A shallow curve indicates that the library has reached complexity saturation and further sequencing would likely not add further unique reads. The dashed line shows a perfectly complex library where total reads = unique reads.
Note that these are predictive numbers only, not absolute. The MultiQC plot can sometimes give extreme sequencing depth on the X axis - click and drag from the left side of the plot to zoom in on more realistic numbers.
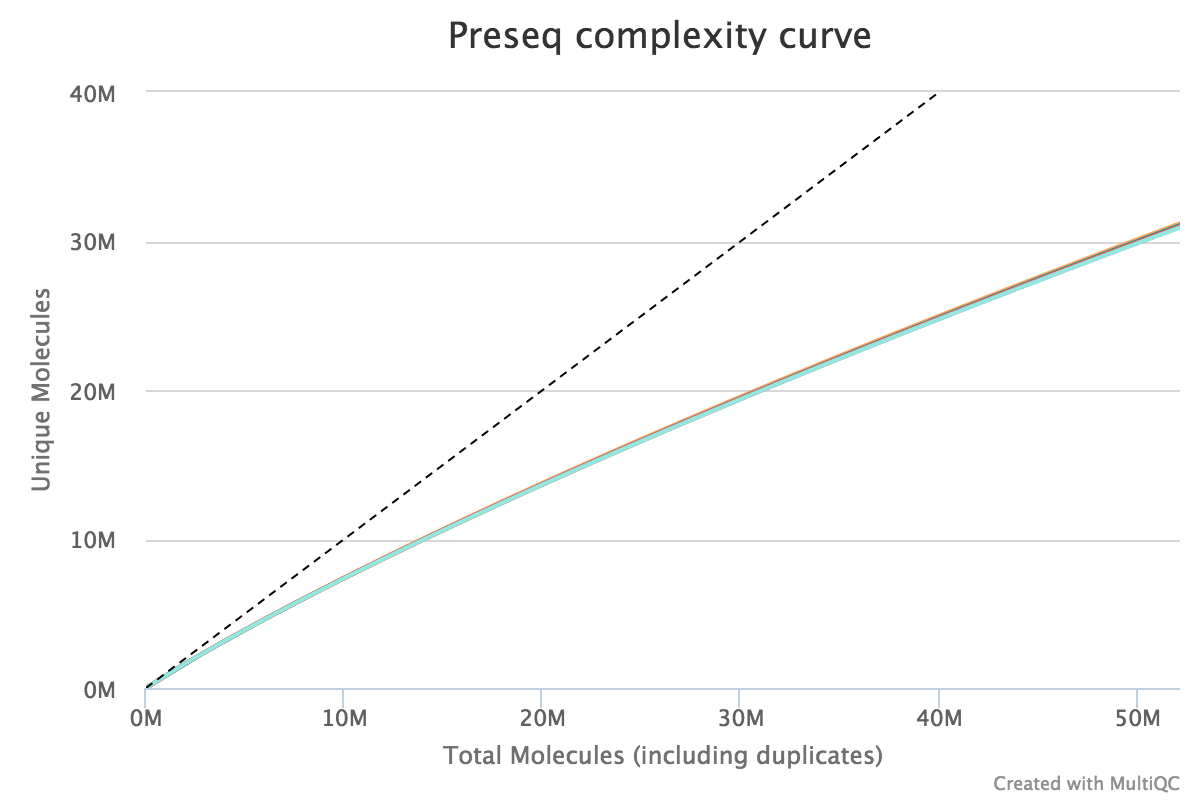
Output directory: results/preseq
sample_ccurve.txt- This file contains plot values for the complexity curve, plotted in the MultiQC report.
featureCounts
featureCounts from the subread package summarises the read distribution over genomic features such as genes, exons, promotors, gene bodies, genomic bins and chromosomal locations. RNA reads should mostly overlap genes, so be assigned.
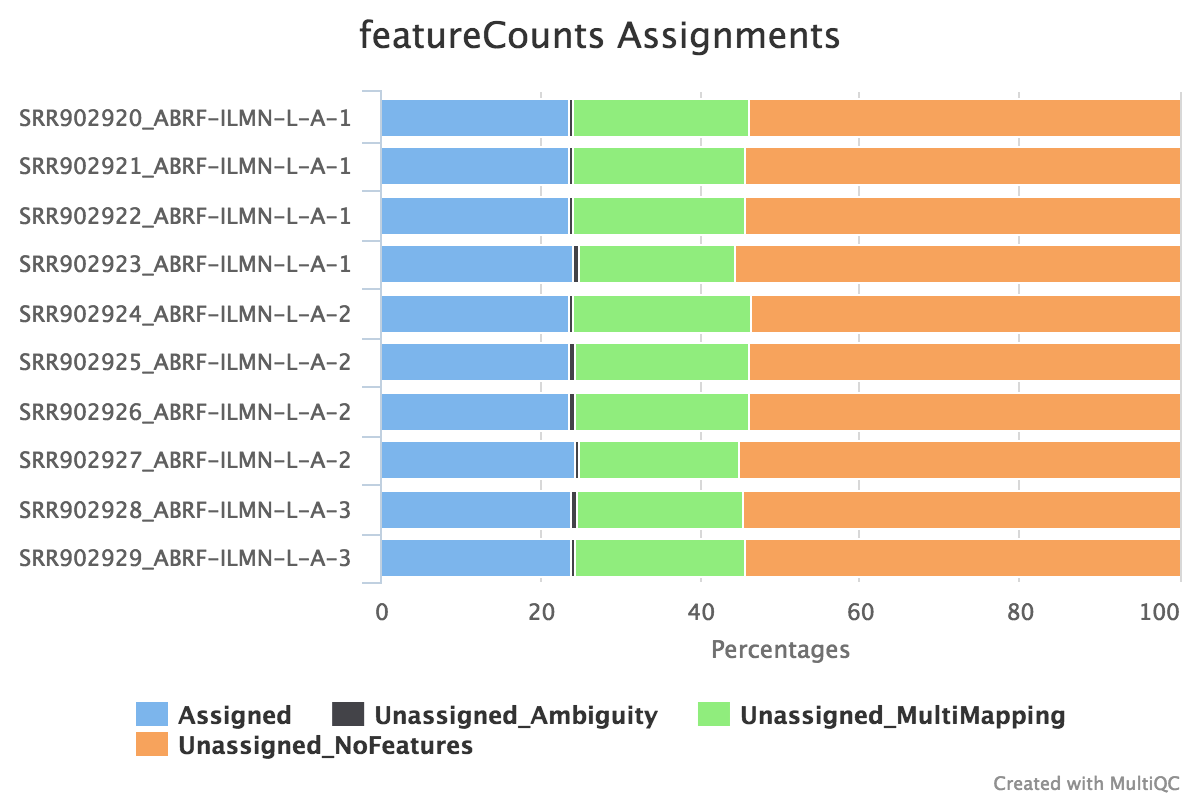
We also use featureCounts to count overlaps with different classes of features. This gives a good idea of where aligned reads are ending up and can show potential problems such as rRNA contamination.
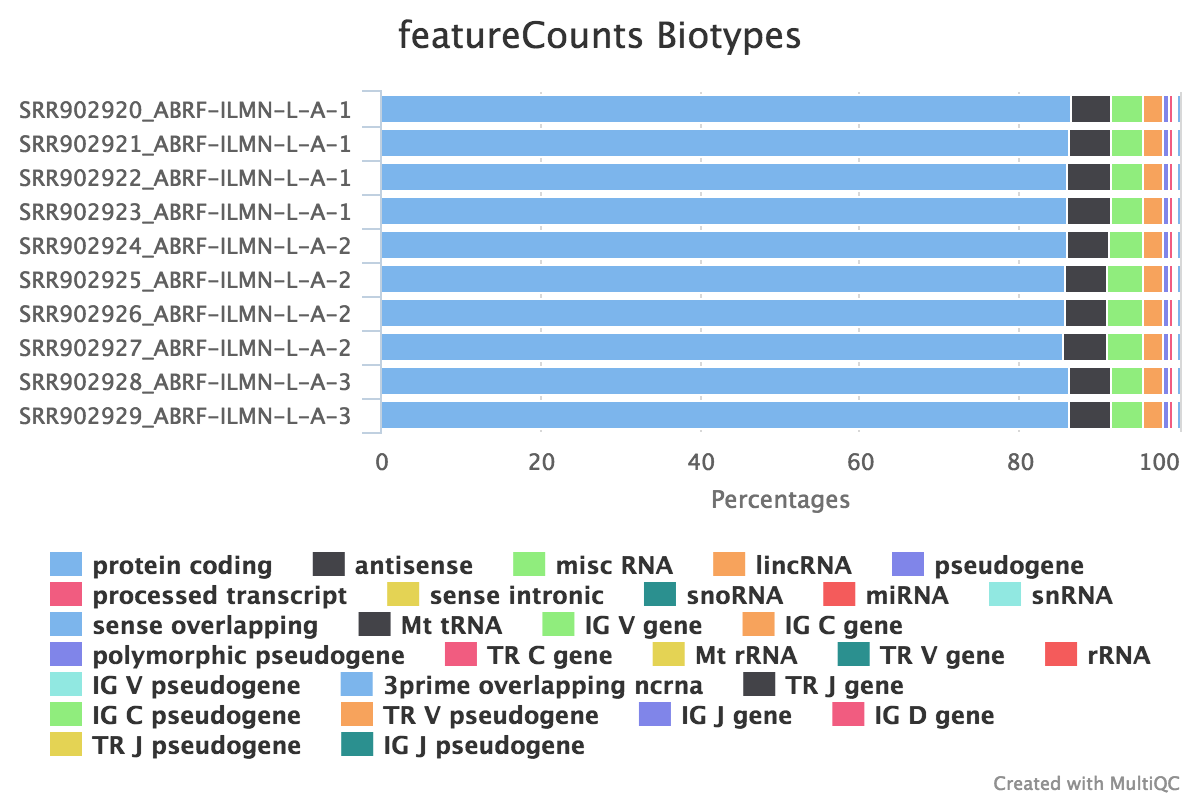
Output directory: results/featureCounts
Sample.bam_biotype_counts.txt- Read counts for the different gene biotypes that featureCounts distinguishes.
Sample.featureCounts.txt- Read the counts for each gene provided in the reference
gtffile
- Read the counts for each gene provided in the reference
Sample.featureCounts.txt.summary- Summary file, containing statistics about the counts
Salmon
Salmon from Ocean Genomics quasi-maps and quantifies expression relative to the transcriptome.
Output directory: results/salmon
Sample/quant.sf- Read counts for the different transcripts.
Sample/quant.genes.sf- Read the counts for each gene provided in the reference
gtffile
- Read the counts for each gene provided in the reference
Sample/logs- Summary file with information about the process
unaligned/- Contains a list of unmapped reads that can be used to generate a FastQ of unmapped reads for downstream analysis.
tximport
tximport imports transcript-level abundance, estimated counts and transcript lengths, and summarizes into matrices for use with downstream gene-level analysis packages. Average transcript length, weighted by sample-specific transcript abundance estimates, is provided as a matrix which can be used as an offset for different expression of gene-level counts.
Output directory: results/salmon
salmon_merged_transcript_tpm.csv- TPM counts for the different transcripts.
salmon_merged_gene_tpm.csv- TPM counts for the different genes.
salmon_merged_transcript_counts.csv- estimated counts for the different transcripts.
salmon_merged_gene_counts.csv- estimated counts for the different genes.
tx2gene.csv- CSV file with transcript and genes (
params.fc_group_features) and extra name (params.fc_extra_attributes) in each column.
- CSV file with transcript and genes (
se.rds- RDS object to be loaded in R that contains a SummarizedExperiment with the TPM (
abundance), estimated counts (counts) and transcript length (length) in the assays slot for transcripts.
- RDS object to be loaded in R that contains a SummarizedExperiment with the TPM (
gse.rds- RDS object to be loaded in R that contains a SummarizedExperiment with the TPM (
abundance), estimated counts (counts) and transcript length (length) in the assays slot for genes.
- RDS object to be loaded in R that contains a SummarizedExperiment with the TPM (
Index files
Output directory: results/reference_genome/salmon_index
duplicate_clusters.tsv- Stores which transcripts are duplicates of one another
hash.binheader.json- Information about k-mer size, uniquely identifying hashes for the reference
indexing.log- Time log for creating transcriptome index
quasi_index.log- Step-by-step log for making transcriptome index
refInfo.json- Information about file used for the reference
rsd.binsa.bintxpInfo.binversionInfo.json- Salmon and indexing version sed to make the index
Quantification output
Output directory: results/salmon
aux_info/- Auxiliary info e.g. versions and number of mapped reads
cmd_info.json- Information about the Salmon quantification command, version, and options
lib_format_counts.json- Number of fragments assigned, unassigned and incompatible
libParams/- Contains the file
flenDist.txtfor the fragment length distribution
- Contains the file
logs/- Contains the file
salmon_quant.loggiving a record of Salmon’s quantification
- Contains the file
quant.sf- Transcript-level quantification of the sample, including gene length, effective length, TPM, and number of reads
quant.genes.sf- Gene-level quantification of the sample, including gene length, effective length, TPM, and number of reads
Sample.transcript.tpm.txt- Subset of
quant.sf, only containing the transcript id and TPM values
- Subset of
Sample.gene.tpm.txt- Subset of
quant.genes.sf, only containing the gene id and TPM values
- Subset of
StringTie
StringTie assembles RNA-Seq alignments into potential transcripts. It assembles and quantitates full-length transcripts representing multiple splice variants for each gene locus.
StringTie outputs FPKM metrics for genes and transcripts as well as the transcript features that it generates.
Output directory: results/stringtie
<sample>_Aligned.sortedByCoord.out.bam.gene_abund.txt- Gene aboundances, FPKM values
<sample>_Aligned.sortedByCoord.out.bam_transcripts.gtf- This
.gtffile contains all of the assembled transcipts from StringTie
- This
<sample>_Aligned.sortedByCoord.out.bam.cov_refs.gtf- This
.gtffile contains the transcripts that are fully covered by reads.
- This
Sample Correlation
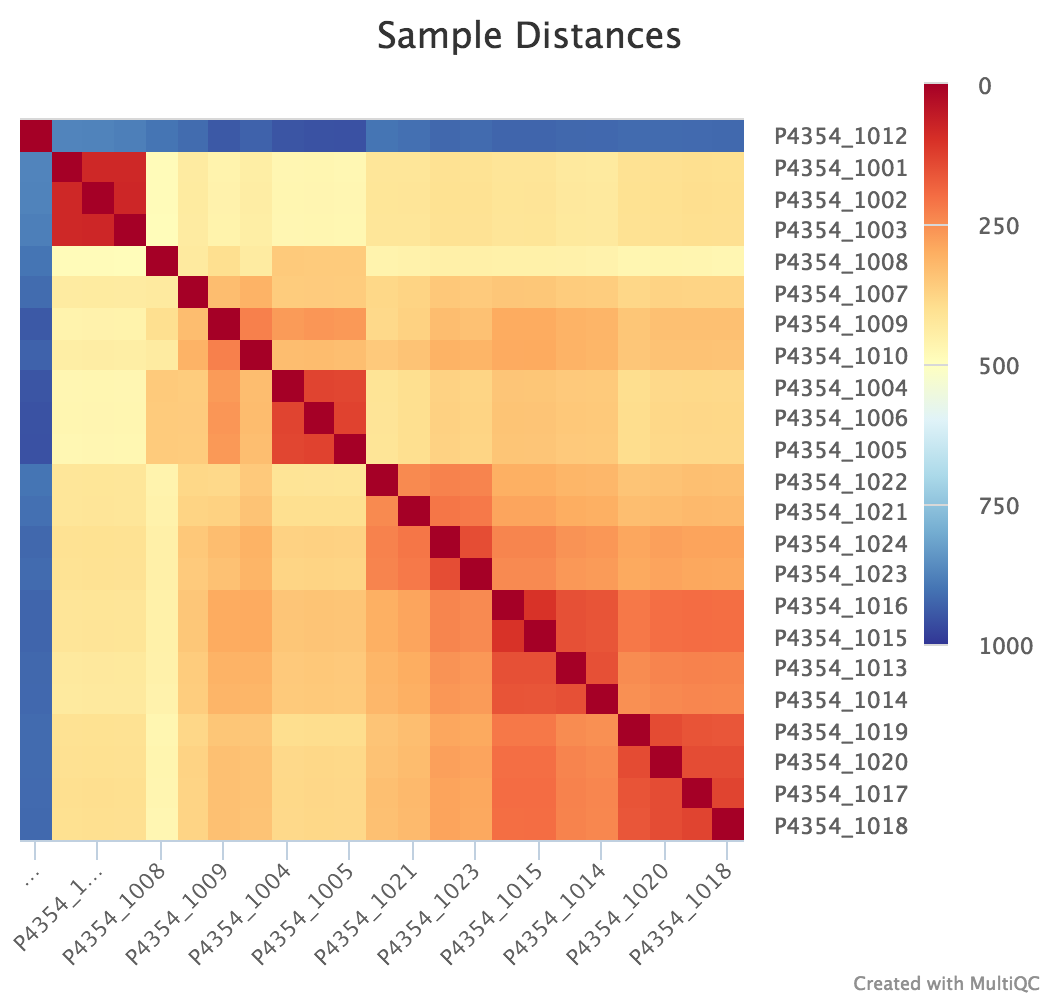
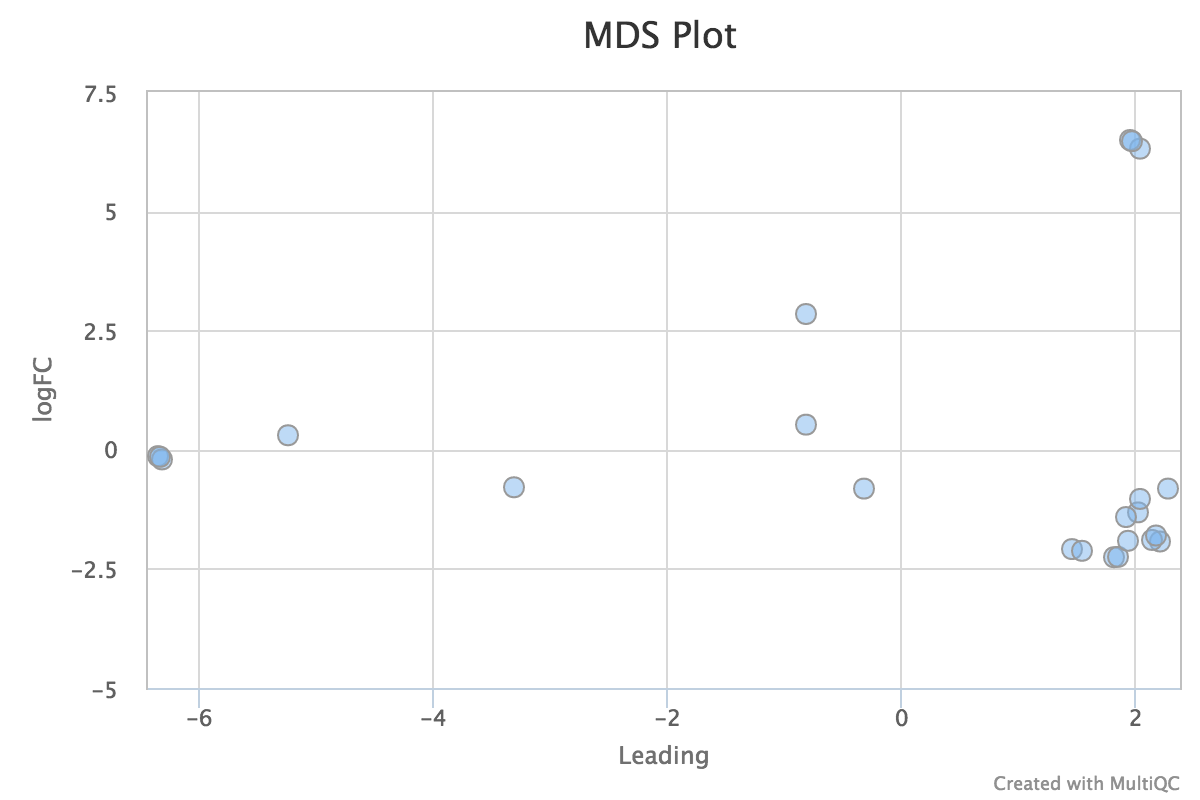
edgeR is a Bioconductor package for R used for RNA-seq data analysis. The script included in the pipeline uses edgeR to normalise read counts and create a heatmap showing Pearson’s correlation and a dendrogram showing pairwise Euclidean distances between the samples in the experiment. It also creates a 2D MDS scatter plot showing sample grouping. These help to show sample similarity and can reveal batch effects and sample groupings.
Heatmap:
MDS plot:
Output directory: results/sample_correlation
edgeR_MDS_plot.pdf- MDS scatter plot showing sample similarity
edgeR_MDS_distance_matrix.csv- Distance matrix containing raw data from MDS analysis
edgeR_MDS_Aplot_coordinates_mqc.csv- Scatter plot coordinates from MDS plot, used for MultiQC report
log2CPM_sample_distances_dendrogram.pdf- Dendrogram showing the Euclidean distance between your samples
log2CPM_sample_correlation_heatmap.pdf- Heatmap showing the Pearsons correlation between your samples
log2CPM_sample_correlation_mqc.csv- Raw data from Pearsons correlation heatmap, used for MultiQC report
MultiQC
MultiQC is a visualisation tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available in within the report data directory.
The pipeline has special steps which allow the software versions used to be reported in the MultiQC output for future traceability.
Output directory: results/multiqc
Project_multiqc_report.html- MultiQC report - a standalone HTML file that can be viewed in your web browser
Project_multiqc_data/- Directory containing parsed statistics from the different tools used in the pipeline
For more information about how to use MultiQC reports, see http://multiqc.info