nf-core/mag
Assembly and binning of metagenomes
2.4.0). The latest
stable release is
5.4.1
.
Introduction
nf-core/mag is a bioinformatics best-practise analysis pipeline for assembly, binning and annotation of metagenomes.
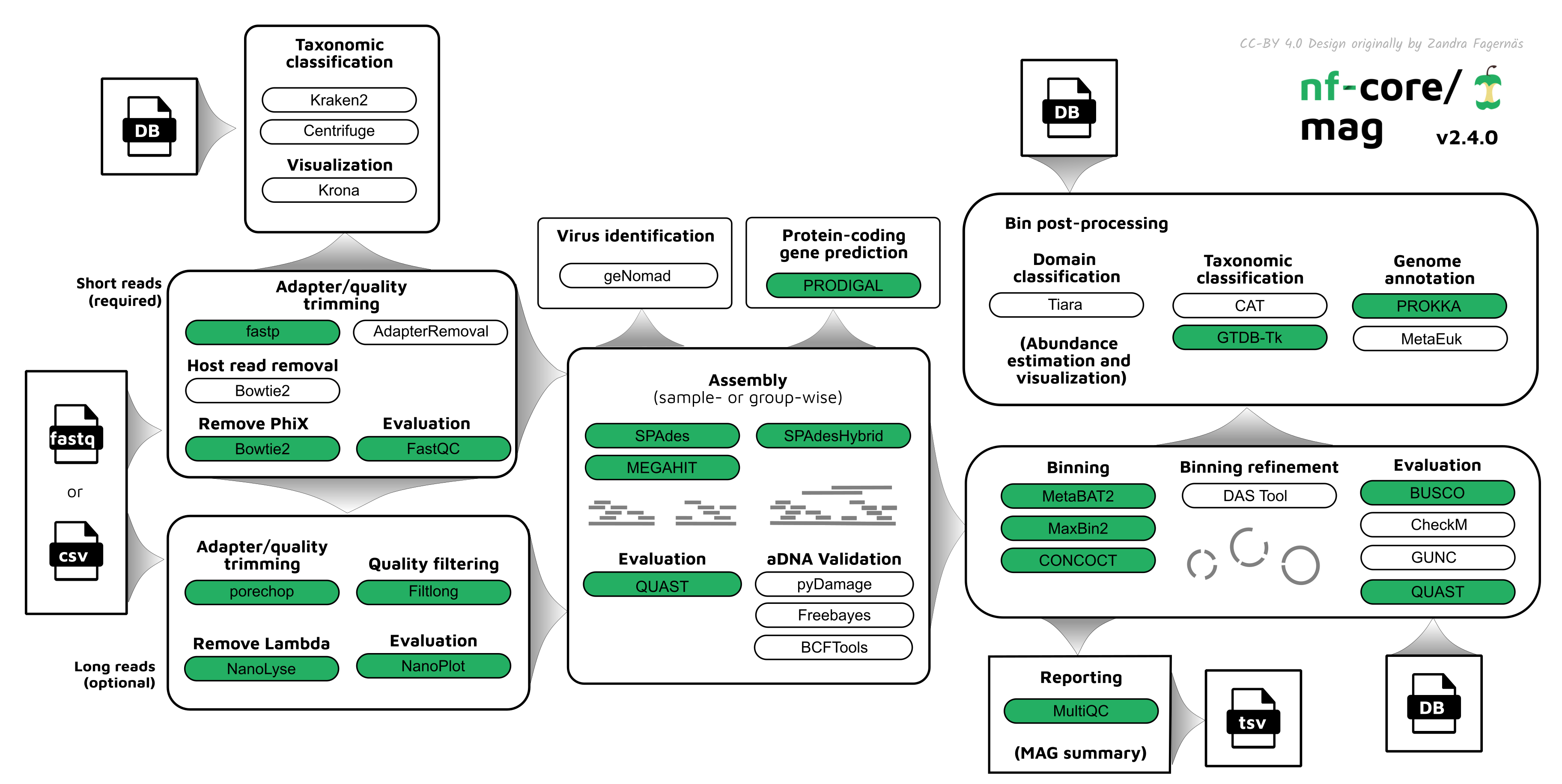
Pipeline summary
By default, the pipeline currently performs the following: it supports both short and long reads, quality trims the reads and adapters with fastp and Porechop, and performs basic QC with FastQC, and merge multiple sequencing runs.
The pipeline then:
- assigns taxonomy to reads using Centrifuge and/or Kraken2
- performs assembly using MEGAHIT and SPAdes, and checks their quality using Quast
- (optionally) performs ancient DNA assembly validation using PyDamage and contig consensus sequence recalling with Freebayes and BCFtools
- predicts protein-coding genes for the assemblies using Prodigal, and bins with Prokka and optionally MetaEuk
- performs metagenome binning using MetaBAT2, MaxBin2, and/or with CONCOCT, and checks the quality of the genome bins using Busco, or CheckM, and optionally GUNC.
- Performs ancient DNA validation and repair with pyDamage and freebayes
- optionally refines bins with DAS Tool
- assigns taxonomy to bins using GTDB-Tk and/or CAT and optionally identifies viruses in assemblies using geNomad, or Eukaryotes with Tiara
Furthermore, the pipeline creates various reports in the results directory specified, including a MultiQC report summarizing some of the findings and software versions.
Usage
Note If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with
-profile testbefore running the workflow on actual data.
nextflow run nf-core/mag -profile <docker/singularity/podman/shifter/charliecloud/conda/institute> --input '*_R{1,2}.fastq.gz' --outdir <OUTDIR>or
nextflow run nf-core/mag -profile <docker/singularity/podman/shifter/charliecloud/conda/institute> --input samplesheet.csv --outdir <OUTDIR>See usage docs and parameter docs for all of the available options when running the pipeline.
Warning: Please provide pipeline parameters via the CLI or Nextflow
-params-fileoption. Custom config files including those provided by the-cNextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Group-wise co-assembly and co-abundance computation
Each sample has an associated group ID (see input specifications). This group information can be used for group-wise co-assembly with MEGAHIT or SPAdes and/or to compute co-abundances for the binning step with MetaBAT2. By default, group-wise co-assembly is disabled, while the computation of group-wise co-abundances is enabled. For more information about how this group information can be used see the documentation for the parameters --coassemble_group and --binning_map_mode.
When group-wise co-assembly is enabled, SPAdes is run on accordingly pooled read files, since metaSPAdes does not yet allow the input of multiple samples or libraries. In contrast, MEGAHIT is run for each group while supplying lists of the individual readfiles.
Credits
nf-core/mag was written by Hadrien Gourlé at SLU, Daniel Straub and Sabrina Krakau at the Quantitative Biology Center (QBiC). James A. Fellows Yates and Maxime Borry at the Max Planck Institute for Evolutionary Anthropology joined in version 2.2.0.
Long read processing was inspired by caspargross/HybridAssembly written by Caspar Gross @caspargross
We thank the following people for their extensive assistance in the development of this pipeline:
- Alexander Peltzer
- Antonia Schuster
- Phil Ewels
- Gisela Gabernet
- Harshil Patel
- Johannes Alneberg
- Maxime Garcia
- Michael L Heuer
- Alex Hübner
- Jim Downie
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #mag channel (you can join with this invite).
Citations
If you use nf-core/mag for your analysis, please cite the preprint as follows:
nf-core/mag: a best-practice pipeline for metagenome hybrid assembly and binning
Sabrina Krakau, Daniel Straub, Hadrien Gourlé, Gisela Gabernet, Sven Nahnsen.
NAR Genom Bioinform. 2022 Feb 2;4(1):lqac007. doi: 10.1093/nargab/lqac007.
additionally you can cite the pipeline directly with the following doi: 10.5281/zenodo.3589527
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.