Creating pipelines with nf-core
Material originally written for the Nextflow & nf-core training, October 2022.
Introduction
Welcome to the nf-core section of the Nextflow & nf-core training course! You can follow the commands and examples covered within this training in the document below.
Using GitPod
As with the rest of the Nextflow training up until this point, we will be using GitPod for this training. We will use a different GitPod environment in order to get the very latest releases of the nf-core tools. To launch GitPod, follow the link:
Working in an empty directory
To work with a clean directory, you can do the following:
-
In the terminal, make a new directory to work in:
cd ~ mkdir training cd training -
In the menu top left, select File > Open Folder (^ ⇧ O or ^ O)
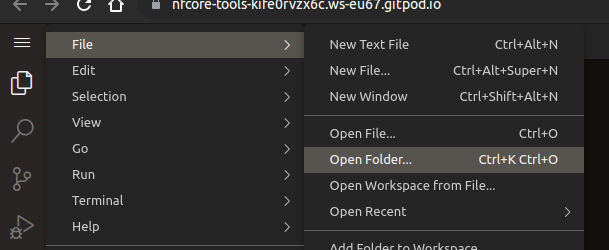
-
Enter
/home/gitpod/training -
GitPod will probably reload the browser tab
-
The file explorer on the left should now have an expandable box with the title
TRAINING
💡 ♻️ As you create new files you should see the explorer populate with them. If you don’t see a file you expect, try hovering over the
TRAININGsection title and clicking the ⟳ icon that appears to refresh the file list.
Local installation
If you prefer, you can install the nf-core/tools package locally.
It is a Python package, available via the Python Package Index (pip) and bioconda (conda / mamba).
There is a docker container available, however for the purposes of this tutorial it not recommended to use this.
To install from PyPI:
pip install nf-coreTo get the very latest development version of the code:
pip install --upgrade --force-reinstall git+https://github.com/nf-core/tools.git@devIf using conda, first set up Bioconda as described in the bioconda docs (especially setting the channel order) and then install nf-core:
conda install nf-coreFirst run of nf-core
Whether running using GitPod or locally, you can confirm that nf-core is correctly installed by viewing the command line help:
nf-core --helpYou should get something that looks like the following output:

The first set of subcommands are typically useful for running pipelines, the second are for developing pipelines.
You can try out some commands, for example listing available nf-core pipelines:

In this tutorial we will focus on creating a pipeline, but please do look at the functionality that nf-core/tools provides to you as a user - especially nf-core pipelines launch and nf-core pipelines download.
Creating a pipeline
To get started with your new pipeline, run the create command:
nf-core createAlthough you can provide options on the command line, it’s easiest to use the interactive prompts.

Follow the instructions and you should see a new pipeline appear in your file explorer.
Let’s move into the new pipeline directory in the terminal:
# NB: The path will vary according to what you called your pipeline!
cd nf-core-demo/Pipeline git repo
The nf-core pipelines create command has made a fully fledged pipeline for you.
Before getting too carried away looking at all of the files, note that it has also initiated a git repository:

It’s actually created three branches for you:

Each have the same initial commit, with the vanilla template:

This is important, because this shared git history with unmodified nf-core template in the TEMPLATE branch is how the nf-core automated template synchronisation works (see the docs for more details).
The main thing to remember with this is that:
- When creating a new repository on https://github.com or equivalent, don’t initialise it - leave it bare and push everything from your local clone
- Develop your code on either the
masterordevbranches and leaveTEMPLATEalone.
Testing the new pipeline
The new pipeline should run with Nextflow, right out of the box. Let’s try:
cd ../
nextflow run nf-core-demo/ -profile test,docker --outdir test_results
Sure enough, our new pipeline has run FastQC and MultiQC on a selection of test data.
Customising the template
In many of the files generated by the nf-core template, you’ll find code comments that look like this:
// TODO nf-core: Do something hereThese are markers to help you get started with customising the template code as you write your pipeline. Editor tools such as Todo tree help you easily navigate these and work your way through them.
Linting your pipeline
Customising the template is part of writing your new pipeline. However, not all files should be edited - indeed, nf-core strives to promote standardisation amongst pipelines.
To try to keep pipelines up to date and using the same code where possible, we have an automated code linting tool for nf-core pipelines.
Running nf-core pipelines lint will run a comprehensive test suite against your pipeline:
cd nf-core-demo/
nf-core lintLinting tests can have one of four statuses: pass, ignore, warn or fail.
For example, at first you will see a large number of warnings about TODO comments, letting you know that you haven’t finished setting up your new pipeline:

Warnings are ok at this stage, but should be cleared up before a pipeline release.
Failures are more serious however, and will typically prevent pull-requests from being merged.
For example, if you edit CODE_OF_CONDUCT.md, which should match the template, you’ll get a pipeline lint test failure:
echo "Edited" >> CODE_OF_CONDUCT.md
nf-core lint
Continuous integration testing
Whilst it’s helpful to be able to run the nf-core lint tests locally, their real strength is the combination with CI (continuous integration) testing.
By default, nf-core pipelines are configured to run CI tests on GitHub every time you push to the repo or open a pull request.
The same nf-core pipelines lint command runs on your code on the automated machines.
If there are any failures, they will be reported with a ❌ and you will not be able to merge the pull-request until you push more commits that fix those failures.
These automated tests allow us to maintain code quality in a scalable and standardised way across the community.
Configuring linting
In some special cases (especially using the nf-core template outside of the nf-core community) you may find that you want to ignore certain linting failures.
To do so, edit .nf-core.yml in the root of the repository.
For example, to ignore the tests triggering warnings in the example above, you can add:
lint:
pipeline_todos: false
files_unchanged:
- CODE_OF_CONDUCT.mdPlease see the linting documentation for specific details of how to configure different tests.
Nextflow Schema
All nf-core pipelines can be run with --help to see usage instructions.
We can try this with the demo pipeline that we just created:
cd ../
nextflow run nf-core-demo/ --help
Here we get a rich help output, with command line parameter variable types, and help text.
However, the Nextflow syntax in nextflow.config does not allow for this kind of rich metadata natively.
To provide this, we created a standard for describing pipeline parameters in a file in the pipeline root called nextflow_schema.json.
These files are written using JSON Schema, making them compatible with many other tools and validation libraries.
By describing our workflow parameters in this file we can do a lot of new things:
- Generate command line help text and web based documentation pages
- Validate pipeline inputs
- Create rich pipeline launch interfaces
Indeed, if you try to run the new pipeline without the required --outdir parameter, you will quickly get an error:
nextflow run nf-core-demo/ -profile test,docker
Working with Nextflow schema
If you peek inside the nextflow_schema.json file you will see that it is quite an intimidating thing.
The file is large and complex, and very easy to break if edited manually.
Thankfully, we provide a user-friendly tool for editing this file: nf-core pipelines schema build.
To see this in action, let’s add some new parameters to nextflow.config:
params {
demo = 'param-value-default'
foo = null
bar = false
baz = 12
// rest of the config file..Then run nf-core pipelines schema build:
cd nf-core-demo/
nf-core schema buildThe CLI tool should then prompt you to add each new parameter:
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.5.1 - https://nf-co.re
INFO [✓] Default parameters match schema validation
INFO [✓] Pipeline schema looks valid (found 28 params)
✨ Found 'params.demo' in the pipeline config, but not in the schema. Add to pipeline schema? [y/n]: y
✨ Found 'params.foo' in the pipeline config, but not in the schema. Add to pipeline schema? [y/n]: y
✨ Found 'params.bar' in the pipeline config, but not in the schema. Add to pipeline schema? [y/n]: y
✨ Found 'params.baz' in the pipeline config, but not in the schema. Add to pipeline schema? [y/n]: y
INFO Writing schema with 32 params: './nextflow_schema.json'
🚀 Launch web builder for customisation and editing? [y/n]: ySelect y on the final prompt to launch a web browser to edit your schema graphically.
⚠️ When using GitPod, a slightly odd thing can happen at this point.
GitPod may launch the schema editor with a terminal-based web browser called
lynx. You’ll see the terminal output suddenly fill with a text-based representation of the nf-core website.
- Press Q followed by Y to confirm to exit
lyx.- You should still see nf-core/tools running on the command line.
- Copy the URL that was printed to the terminal and open this in a new browser tab (or alt + click it).
Here in the schema editor you can edit:
- Description and help text
- Type (string / boolean / integer etc)
- Grouping of parameters
- Whether a parameter is required, or hidden from help by default
- Enumerated values (choose from a list)
- Min / max values for numeric types
- Regular expressions for validation
- Special formats for strings, such as
file-path - Additional fields for files such as
mime-type
nf-core modules
Using the nf-core-demo pipeline that we created above, let’s see how we can add our own modules to the pipeline.
Add a process to the main workflow
Let’s add a simple Nextflow process to our pipeline:
process ECHO_READS {
debug true
input:
tuple val(meta), path(reads)
script:
"""
echo ${reads}
"""
}Paste the process in workflows/demo.nf:
<TRUNCATED>
/*
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
RUN MAIN WORKFLOW
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
*/
<<<<<<<<<< PASTE HERE >>>>>>>>>>
// Info required for completion email and summary
def multiqc_report = []
workflow DEMO {
ch_versions = Channel.empty()
<TRUNCATED>Write some code to invoke the process above:
//
// MODULE: Echo reads
//
ECHO_READS (
ch_samplesheet
)and call it in the main workflow definition after FASTQC:
<TRUNCATED>
//
// MODULE: Run FastQC
//
FASTQC (
ch_samplesheet
)
ch_versions = ch_versions.mix(FASTQC.out.versions.first())
<<<<<<<<<< PASTE HERE >>>>>>>>>>
<TRUNCATED>Let’s re-run the pipeline and test it. The new process will just print to screen the file names for the FastQ files that are used for the test dataset with this workflow. Add -resume to the end of the command to ensure that previously run and successful tasks are cached.
$ nextflow run . -profile test,docker --outdir test_results -resume
N E X T F L O W ~ version 22.08.2-edge
Launching `./main.nf` [voluminous_meucci] DSL2 - revision: 282d7ef11e
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo v1.0dev
------------------------------------------------------
Core Nextflow options
runName : voluminous_meucci
containerEngine : docker
launchDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo
workDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo/work
projectDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo
userName : harshil
profile : test,docker
configFiles : nf-core/training/2022_10_04_nf_core_training/nf-core-demo/nextflow.config
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : test_results
Reference genome options
genome : R64-1-1
fasta : s3://ngi-igenomes/igenomes/Saccharomyces_cerevisiae/Ensembl/R64-1-1/Sequence/WholeGenomeFasta/genome.fa
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Max job request options
max_cpus : 2
max_memory : 6.GB
max_time : 6.h
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
If you use nf-core/demo for your analysis please cite:
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
------------------------------------------------------
executor > local (6)
[e9/3a447d] process > NFCORE_DEMO:DEMO:INPUT_CHECK:SAMPLESHEET_CHECK (samplesheet_test_illumina_amplicon.csv) [100%] 1 of 1, cached: 1 ✔
[2e/60807c] process > NFCORE_DEMO:DEMO:FASTQC (SAMPLE2_PE_T1) [100%] 4 of 4, cached: 4 ✔
[4b/0c4607] process > NFCORE_DEMO:DEMO:ECHO_READS (4) [100%] 4 of 4 ✔
[ab/a41ccf] process > NFCORE_DEMO:DEMO:CUSTOM_DUMPSOFTWAREVERSIONS (1) [100%] 1 of 1 ✔
[b6/4cea13] process > NFCORE_DEMO:DEMO:MULTIQC [100%] 1 of 1 ✔
sample1_R1.fastq.gz sample1_R2.fastq.gz
sample1_R1.fastq.gz
sample2_R1.fastq.gz sample2_R2.fastq.gz
sample2_R1.fastq.gz
-[nf-core/demo] Pipeline completed successfully-Add a local module to the pipeline
Create a new file called echo_reads.nf in modules/local/. Cut and paste the process definition into this file and save it.
Now import it in workflows/demo.nf by pasting the snippet below with all of the other include statements in that file:
include { ECHO_READS } from '../modules/local/echo_reads'Let’s re-run the pipeline and test it again to make sure it is working:
$ nextflow run . -profile test,docker --outdir test_results -resume
N E X T F L O W ~ version 22.08.2-edge
Launching `./main.nf` [shrivelled_visvesvaraya] DSL2 - revision: 282d7ef11e
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo v1.0dev
------------------------------------------------------
Core Nextflow options
runName : shrivelled_visvesvaraya
containerEngine : docker
launchDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo
workDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo/work
projectDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo
userName : harshil
profile : test,docker
configFiles : nf-core/training/2022_10_04_nf_core_training/nf-core-demo/nextflow.config
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : test_results
Reference genome options
genome : R64-1-1
fasta : s3://ngi-igenomes/igenomes/Saccharomyces_cerevisiae/Ensembl/R64-1-1/Sequence/WholeGenomeFasta/genome.fa
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Max job request options
max_cpus : 2
max_memory : 6.GB
max_time : 6.h
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
If you use nf-core/demo for your analysis please cite:
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
------------------------------------------------------
executor > local (2)
[e9/3a447d] process > NFCORE_DEMO:DEMO:INPUT_CHECK:SAMPLESHEET_CHECK (samplesheet_test_illumina_amplicon.csv) [100%] 1 of 1, cached: 1 ✔
[3f/90b51e] process > NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE_T2) [100%] 4 of 4, cached: 4 ✔
[0d/d24e60] process > NFCORE_DEMO:DEMO:ECHO_READS (2) [100%] 4 of 4, cached: 4 ✔
[fa/fe4aad] process > NFCORE_DEMO:DEMO:CUSTOM_DUMPSOFTWAREVERSIONS (1) [100%] 1 of 1 ✔
[0c/9b887d] process > NFCORE_DEMO:DEMO:MULTIQC [100%] 1 of 1 ✔
sample1_R1.fastq.gz
sample1_R1.fastq.gz sample1_R2.fastq.gz
sample2_R1.fastq.gz
sample2_R1.fastq.gz sample2_R2.fastq.gz
-[nf-core/demo] Pipeline completed successfully-You can now include the same process as many times as you like in the pipeline which is one of the primary strengths of the Nextflow DSL2 syntax:
include { ECHO_READS as ECHO_READS_ONCE } from '../modules/local/echo_reads'
include { ECHO_READS as ECHO_READS_TWICE } from '../modules/local/echo_reads'Try to change the workflow to call the processes above which should print the FastQ file names twice to screen instead of once.
List modules in pipeline
The nf-core pipeline template comes with a few nf-core/modules pre-installed. You can list these with the command below:
$ nf-core modules list local
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
INFO Modules installed in '.': list.py:136
┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┓
┃ Module Name ┃ Repository ┃ Version SHA ┃ Message ┃ Date ┃
┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━┩
│ custom/dumpsoftwareversions │ https://github.com/nf-core/modules.git │ 5e34754d42cd2d5d248ca8673c0a53cdf5624905 │ Restructure nf-core/modules repo (#2141) │ 2022-10-04 │
│ fastqc │ https://github.com/nf-core/modules.git │ 5e34754d42cd2d5d248ca8673c0a53cdf5624905 │ Restructure nf-core/modules repo (#2141) │ 2022-10-04 │
│ multiqc │ https://github.com/nf-core/modules.git │ 5e34754d42cd2d5d248ca8673c0a53cdf5624905 │ Restructure nf-core/modules repo (#2141) │ 2022-10-04 │
└─────────────────────────────┴────────────────────────────────────────┴──────────────────────────────────────────┴──────────────────────────────────────────┴────────────┘These version hashes and repository information for the source of the modules are tracked in the modules.json file in the root of the repo. This file will automatically be updated by nf-core/tools when you create, remove, update modules.
Update modules in pipeline
Let’s see if all of our modules are up-to-date:
$ nf-core modules update
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
? Update all modules or a single named module? All modules
? Do you want to view diffs of the proposed changes? No previews, just update everything
INFO 'modules/nf-core/custom/dumpsoftwareversions' is already up to date update.py:162
INFO 'modules/nf-core/fastqc' is already up to date update.py:162
INFO 'modules/nf-core/multiqc' is already up to date update.py:162
INFO Updates complete ✨Nothing to see here!
List remote modules on nf-core/modules
You can list all of the modules available on nf-core/modules via the command below but we have added search functionality to the nf-core website to do this too!
$ nf-core modules list remote
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
INFO Modules available from https://github.com/nf-core/modules.git (master): list.py:131
┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓
┃ Module Name ┃
┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩
│ abacas │
│ abricate/run │
│ abricate/summary │
│ adapterremoval │
│ adapterremovalfixprefix │
│ agrvate │
│ allelecounter │
│ ampir │
│ amplify/predict │
│ amps │
│ amrfinderplus/run │
│ amrfinderplus/update │
│ angsd/docounts │
│ antismash/antismashlite │
│ antismash/antismashlitedownloaddatabases │
│ ariba/getref │
│ ariba/run │
│ arriba │
│ artic/guppyplex │
│ artic/minion │
│ ascat │
│ assemblyscan │
│ ataqv/ataqv │
│ ataqv/mkarv │
│ atlas/call │
│ atlas/pmd │
<TRUNCATED>Install a module from nf-core/modules
$ nf-core modules install
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.7.2 - https://nf-co.re
? Tool name: fastp
INFO Installing 'fastp' install.py:128
INFO Include statement: include { FASTP } from '../modules/nf-core/fastp/main' install.py:137Let’s install the FASTP module into the pipeline and fetch it’s key information including input and output channel definitions:
$ nf-core modules info fastp
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
╭─ Module: fastp ────────────────────────────────────────────────────────────────────────────────────────────────────────╮
│ 🌐 Repository: https://github.com/nf-core/modules.git │
│ 🔧 Tools: fastp │
│ 📖 Description: Perform adapter/quality trimming on sequencing reads │
╰─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────╯
╷ ╷
📥 Inputs │Description │Pattern
╺━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┿━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┿━━━━━━━╸
meta (map) │Groovy Map containing sample information. Use 'single_end: true' to specify single │
│ended or interleaved FASTQs. Use 'single_end: false' for paired-end reads. e.g. [ │
│id:'test', single_end:false ] │
╶─────────────────────────────┼───────────────────────────────────────────────────────────────────────────────────┼───────╴
reads (file) │List of input FastQ files of size 1 and 2 for single-end and paired-end data, │
│respectively. If you wish to run interleaved paired-end data, supply as single-end│
│data but with --interleaved_in in your modules.conf's ext.args for the module. │
╶─────────────────────────────┼───────────────────────────────────────────────────────────────────────────────────┼────────────────────╴
adapter_fasta (file) │File in FASTA format containing possible adapters to remove. │*.{fasta,fna,fas,fa}
╶─────────────────────────────┼────────────────────────────────────────────────────────────────────────────────┼───────────────────
save_trimmed_fail (boolean)│Specify true to save files that failed to pass trimming thresholds ending in │
│*.fail.fastq.gz │
╶─────────────────────────────┼────────────────────────────────────────────────────────────────────────────────┼───────╴
save_merged (boolean) │Specify true to save all merged reads to the a file ending in *.merged.fastq.gz │
╵ ╵
╷ ╷
📤 Outputs │Description │ Pattern
╺━━━━━━━━━━━━━━━━━━━━━┿━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┿━━━━━━━━━━━━━━━━━━━╸
meta (map) │Groovy Map containing sample information e.g. [ id:'test', single_end:false ] │
╶─────────────────────┼───────────────────────────────────────────────────────────────────────────────┼───────────────────╴
reads (file) │The trimmed/modified/unmerged fastq reads │ *fastp.fastq.gz
╶─────────────────────┼───────────────────────────────────────────────────────────────────────────────┼───────────────────╴
json (file) │Results in JSON format │ *.json
╶─────────────────────┼───────────────────────────────────────────────────────────────────────────────┼───────────────────╴
html (file) │Results in HTML format │ *.html
╶─────────────────────┼───────────────────────────────────────────────────────────────────────────────┼───────────────────╴
log (file) │fastq log file │ *.log
╶─────────────────────┼───────────────────────────────────────────────────────────────────────────────┼───────────────────╴
versions (file) │File containing software versions │ versions.yml
╶─────────────────────┼───────────────────────────────────────────────────────────────────────────────┼───────────────────╴
reads_fail (file) │Reads the failed the preprocessing │ *fail.fastq.gz
╶─────────────────────┼───────────────────────────────────────────────────────────────────────────────┼───────────────────╴
reads_merged (file)│Reads that were successfully merged │*.{merged.fastq.gz}
╵ ╵
💻 Installation command: nf-core modules install fastp
gitpod /workspace/nf-core-demo (master) $If we inspect the main script for the FASTP module the first input channel looks exactly the same as for the FASTQC module which we already know is working from the tests. We can copy the include statement printed whilst installing the pipeline and paste it in workflows/demo.nf.
We now just need to call the FASTP process in the main workflow. Paste the snippet below just after the call to ECHO_READS_TWICE.
//
// MODULE: Run Fastp trimming
//
FASTP (
ch_samplesheet,
[],
false,
false
)Let’s re-run the pipeline and test it again to make sure it is working:
$ nextflow run . -profile test,docker --outdir test_results -resume
N E X T F L O W ~ version 22.08.2-edge
Launching `./main.nf` [nice_carlsson] DSL2 - revision: 282d7ef11e
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo v1.0dev
------------------------------------------------------
Core Nextflow options
runName : nice_carlsson
containerEngine : docker
launchDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo
workDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo/work
projectDir : nf-core/training/2022_10_04_nf_core_training/nf-core-demo
userName : harshil
profile : test,docker
configFiles : nf-core/training/2022_10_04_nf_core_training/nf-core-demo/nextflow.config
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : test_results
Reference genome options
genome : R64-1-1
fasta : s3://ngi-igenomes/igenomes/Saccharomyces_cerevisiae/Ensembl/R64-1-1/Sequence/WholeGenomeFasta/genome.fa
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Max job request options
max_cpus : 2
max_memory : 6.GB
max_time : 6.h
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
If you use nf-core/demo for your analysis please cite:
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
------------------------------------------------------
executor > local (6)
[e9/3a447d] process > NFCORE_DEMO:DEMO:INPUT_CHECK:SAMPLESHEET_CHECK (samplesheet_test_illumina_amplicon.csv) [100%] 1 of 1, cached: 1 ✔
[2e/60807c] process > NFCORE_DEMO:DEMO:FASTQC (SAMPLE2_PE_T1) [100%] 4 of 4, cached: 4 ✔
[0d/d24e60] process > NFCORE_DEMO:DEMO:ECHO_READS (2) [100%] 4 of 4, cached: 4 ✔
[31/78e46a] process > NFCORE_DEMO:DEMO:FASTP (SAMPLE3_SE_T2) [100%] 4 of 4 ✔
[5a/d1d6ba] process > NFCORE_DEMO:DEMO:CUSTOM_DUMPSOFTWAREVERSIONS (1) [100%] 1 of 1 ✔
[34/f27c22] process > NFCORE_DEMO:DEMO:MULTIQC [100%] 1 of 1 ✔
sample1_R1.fastq.gz
sample1_R1.fastq.gz sample1_R2.fastq.gz
sample2_R1.fastq.gz
sample2_R1.fastq.gz sample2_R2.fastq.gz
-[nf-core/demo] Pipeline completed successfully-Patch a module
Say we want to make a slight change to an existing nf-core/module that is custom to a particular use case, we can create a patch of the module that will be tracked by the commands in nf-core/tools.
Let’s add the snippet below at the top of the script section in the FASTP nf-core module:
<TRUNCATED>
"""
echo "These are my changes"
[ ! -f ${prefix}.fastq.gz ] && ln -sf $reads ${prefix}.fastq.gz
<TRUNCATED>The linting for this module will now fail because the local copy of the module doesn’t match the latest version in nf-core/modules:
$ nf-core modules lint fastp
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
INFO Linting pipeline: '.' __init__.py:202
INFO Linting module: 'fastp' __init__.py:204
╭─ [✗] 1 Module Test Failed ───────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────╮
│ ╷ ╷ │
│ Module name │ File path │ Test message │
│╶──────────────────────────────────────────┼───────────────────────────────┼─────────────────────────────────────────────────────────────────────────────────────────╴│
│ fastp │ modules/nf-core/fastp/main.nf │ Local copy of module does not match remote │
│ ╵ ╵ │
╰──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────╯
╭───────────────────────╮
│ LINT RESULTS SUMMARY │
├───────────────────────┤
│ [✔] 22 Tests Passed │
│ [!] 0 Test Warnings │
│ [✗] 1 Test Failed │
╰───────────────────────╯Fear not! We can just patch the module!
$ nf-core modules patch fastp
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
INFO Changes in module 'nf-core/fastp' modules_differ.py:252
INFO 'modules/nf-core/fastp/meta.yml' is unchanged modules_differ.py:257
INFO Changes in 'fastp/main.nf': modules_differ.py:266
--- modules/nf-core/fastp/main.nf
+++ modules/nf-core/fastp/main.nf
@@ -32,6 +32,8 @@
// Use single ended for interleaved. Add --interleaved_in in config.
if ( task.ext.args?.contains('--interleaved_in') ) {
"""
+ echo "These are my changes"
+
[ ! -f ${prefix}.fastq.gz ] && ln -sf $reads ${prefix}.fastq.gz
fastp \\
INFO Patch file of 'modules/nf-core/fastp' written to 'modules/nf-core/fastp/fastp.diff' patch.py:124The diff is stored in a file:
cat modules/nf-core/fastp/fastp.diff
Changes in module 'nf-core/fastp'
--- modules/nf-core/fastp/main.nf
+++ modules/nf-core/fastp/main.nf
@@ -32,6 +32,8 @@
// Use single ended for interleaved. Add --interleaved_in in config.
if ( task.ext.args?.contains('--interleaved_in') ) {
"""
+ echo "These are my changes"
+
[ ! -f ${prefix}.fastq.gz ] && ln -sf $reads ${prefix}.fastq.gz
fastp \\
************************************************************and the path to this patch file is added to modules.json:
"fastp": {
"branch": "master",
"git_sha": "5e34754d42cd2d5d248ca8673c0a53cdf5624905",
"patch": "modules/nf-core/fastp/fastp.diff"
},Lint all modules
As well as the pipeline template you can lint individual or all modules with a single command:
$ nf-core modules lint --all
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
INFO Linting pipeline: '.' __init__.py:202
╭─ [!] 1 Module Test Warning ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────╮
│ ╷ ╷ │
│ Module name │ File path │ Test message │
│╶──────────────────────────────────────────┼─────────────────────────────────────────────────────┼───────────────────────────────────────────────────────────────────╴│
│ custom/dumpsoftwareversions │ modules/nf-core/custom/dumpsoftwareversions/main.nf │ Process name not used for versions.yml │
│ ╵ ╵ │
╰──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────╯
╭──────────────────────╮
│ LINT RESULTS SUMMARY │
├──────────────────────┤
│ [✔] 89 Tests Passed │
│ [!] 1 Test Warning │
│ [✗] 0 Tests Failed │
╰──────────────────────╯Create a local module
$ nf-core modules create
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.6 - https://nf-co.re
INFO Repository type: pipeline create.py:99
INFO Press enter to use default values (shown in brackets) or type your own responses. ctrl+click underlined text to create.py:103
open links.
Name of tool/subtool: demo/module
WARNING Could not find Conda dependency using the Anaconda API: 'demo' create.py:174
Do you want to enter a different Bioconda package name? [y/n]: n
WARNING Could not find Conda dependency using the Anaconda API: 'demo' create.py:181
Building module without tool software and meta, you will need to enter this information manually.
GitHub Username: (@author): @drpatelh
INFO Provide an appropriate resource label for the process, taken from the nf-core pipeline template. create.py:224
For example: process_single, process_low, process_medium, process_high, process_long
? Process resource label: process_single
INFO Where applicable all sample-specific information e.g. 'id', 'single_end', 'read_group' MUST be provided as an create.py:238
input via a Groovy Map called 'meta'. This information may not be required in some instances, for example
indexing reference genome files.
Will the module require a meta map of sample information? [y/n] (y): y
INFO Created / edited following files: create.py:276
./modules/local/demo/module.nfOpen ./modules/local/demo/module.nf and start customising this to your needs whilst working your way through the extensive TODO comments!
Contribute to nf-core/modules
$ git clone https://github.com/nf-core/modules.git
$ cd modules
$ git checkout -b demo_branch
$ nf-core modules create
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 2.11 - https://nf-co.re
INFO Repository type: modules
INFO Press enter to use default values (shown in brackets) or type your own responses. ctrl+click underlined text to open links.
Name of tool/subtool: test
INFO Using Bioconda package: 'bioconda::test=0.1'
INFO Could not find a Docker/Singularity container (Unexpected response code `204` for
https://api.biocontainers.pro/ga4gh/trs/v2/tools/test/versions/test-0.1)
GitHub Username: (@myuser):
INFO Provide an appropriate resource label for the process, taken from the nf-core pipeline template.
For example: process_single, process_low, process_medium, process_high, process_long
? Process resource label: process_single
INFO Where applicable all sample-specific information e.g. 'id', 'single_end', 'read_group' MUST be provided as an input via a
Groovy Map called 'meta'. This information may not be required in some instances, for example indexing reference genome
files.
Will the module require a meta map of sample information? [y/n] (y):
INFO Created component template: 'test'
INFO Created following files:
modules/nf-core/test/main.nf
modules/nf-core/test/meta.yml
modules/nf-core/test/environment.yml
modules/nf-core/test/tests/tags.yml
modules/nf-core/test/tests/main.nf.testYou will see that when you create a module in a clone of the modules repository more files are added than if you create a local module in the pipeline as we did in the previous section. See the DSL2 modules docs on the nf-core website for further information.